一种1,2,3
‑
三氯丙烷的连续流制备方法
技术领域
1.本发明属于有机合成技术领域,具体涉及一种1,2,3
‑
三氯丙烷的连续流制备方法。
背景技术:
2.1,2,3
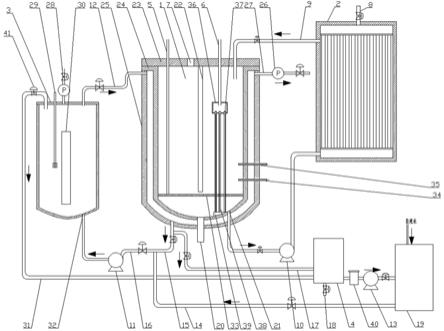
‑
三氯丙烷是重要的化工中间体,其cas号为96
‑
18
‑
4,分子式为c3h5cl3,其结构式为:。
3.1,2,3
‑
三氯丙烷为优良的溶剂,主要用作脱脂剂、去漆剂和电机洗涤用溶剂,还可用于生产三氯丙烯及农药矮壮素和燕麦敌等。其作为第二代烟碱类杀虫剂噻虫嗪、噻虫胺和医药利托那韦合成的关键起始原料,近几年来已逐渐引起了农药研究者们的广泛关注,1,2,3
‑
三氯丙烷产品经裂解反应可制备2,3
‑
二氯丙烯,通过与硫氰酸盐取代反应得2
‑
氯丙烯基异硫氰酸酯,再通氯气氯化可得到2
‑
氯
‑5‑
氯甲基噻唑中间体,该中间体随着烟碱类杀虫剂的快速发展,市场需求量巨大。
4.现有技术中,主要使用α
‑
氯丙烯(3
‑
氯丙烯)氯化法合成1,2,3
‑
三氯丙烷,该方法采用间歇釜式反应,最终通过精馏得成品,该方法虽然氯气的转化率可以达到95%,但纯度相对较低,且进料量难以控制。该反应方法生产成本高,反应温度控制难度大,并且产品质量不高,尤其收率较低,副产物多,污水废气处理难度大。
5.另外,de3432720a1提供了一种通过烯丙基氯与磺酰氯的反应制备1,2,3
‑
三氯丙烷的方法,具体为在液相中,在含氮碱、膦和/或氧化膦存在的条件下,在烯丙基氯中加入socl2,在约30℃至约70℃的温度范围内反应制备得到1,2,3
‑
三氯丙烷。该反应需要在无光的条件下进行,反应时间相对较长,反应中产生的so2、hcl气体逸出对环境造成污染,并且需要在减压条件下分离所得产物,甚至需要通过水洗来分离磺化副产物,不利于工业化生产。
6.cn108640811a提供了一种氯丙烯合成制备1,2,3
‑
三氯丙烷的方法,其采用气
‑
液反应的方式向氯丙烯中通入氯气、并通过冰水浴维持反应温度在20
‑
40℃下制备1,2,3
‑
三氯丙烯,但其反应时间较长、氯气消耗量极大且容易逸出,且需要经过碳酸钠水溶液清洗产品至中性并需要使用干燥剂及过滤,在经过复杂的后处理过程精制后的产品纯度至高为96.5%,并不十分令人满意。
7.cn108658722a提供了一种以3
‑
氯丙烯(α
‑
氯丙烯)和氯气为原料,采用气相合成法合成1,2,3
‑
三氯丙烷的方法,该反应需要先在超过45℃的高温下将3
‑
氯丙烯汽化,将汽化后的3
‑
氯丙烯和氮气或其他惰性气体与氯气通入到氯化塔中进行反应,得到的产物汽化后再经过蒸馏得到1,2,3
‑
三氯丙烷。虽然该方法反应为连续化反应,但反应所需大量氮气或其他惰性气体作为保护气体,投入成本高;反应所需氯气相对于液氯更易溢出,对生产人员的身体健康有一定的伤害;且为了保持气相条件,需要维持高温条件,温度过高产生一定的
副反应,副产物难以分离;气
‑
气反应通量小,单位产能受限,设备投入成本高且后期维护费用昂贵,对工人的综合素质要求高,从经济上考虑不适用于大规模工业化生产。
技术实现要素:
8.本发明的目的在于克服现有技术中的不足,提供了一种反应速度快,反应完全,成品含量高,产品收率高,生产产能高并且不存在三废,对环境友好的1,2,3
‑
三氯丙烷的连续流生产工艺。
9.本发明为了实现上述的技术目的,提供如下的技术方案:一种1,2,3
‑
三氯丙烷的连续流制备方法,通过柱塞泵同时将3
‑
氯丙烯和液氯泵入连续流反应装置内制备1,2,3
‑
三氯丙烷,具体反应方程式如下:;进一步地,所述的连续流反应装置为管式反应器;再进一步地,所述的管式反应器为微通道反应器;进一步地,进料方式为连续进料,用柱塞泵同时将3
‑
氯丙烯和液氯泵入;再进一步地,具体为两台柱塞泵同时将3
‑
氯丙烯和液氯泵入,制备得到1,2,3
‑
三氯丙烷;进一步地,所述3
‑
氯丙烯和液氯泵入前通过流量计;再进一步地,所述流量计为质量流量计或体积流量计;进一步地,反应温度为
‑
20~20℃;进一步地,反应温度为
‑
10~10℃;进一步地,反应温度为0~10℃;进一步地,3
‑
氯丙烯和液氯的摩尔比为1:1.0~1.5;进一步地,3
‑
氯丙烯和液氯的摩尔比为1:1.0~1.06;进一步地,催化剂为n,n
‑
二甲基甲酰胺或乙腈中的任一种;进一步地,在上述技术方案中,反应操作为:将原料3
‑
氯丙烯和液氯通过柱塞泵和流量计以定量的流速泵入微通道反应器,控制反应温度为
‑
20~20℃,反应结束后在0~10℃条件下接收反应液,得到1,2,3
‑
三氯丙烷;进一步地,在上述技术方案中,反应操作为:将原料3
‑
氯丙烯和液氯通过柱塞泵和流量计以定量的流速泵入微通道反应器,控制反应温度为
‑
10~10℃,反应结束后在0~10℃条件下接收反应液,得到1,2,3
‑
三氯丙烷;进一步地,在上述技术方案中,反应操作为:将原料3
‑
氯丙烯和液氯通过柱塞泵和流量计以定量的流速泵入微通道反应器,控制反应温度为0~10℃,反应结束后在0~10℃条件下接收反应液,得到1,2,3
‑
三氯丙烷。
10.由于采用了以上的技术,本发明与现有技术相比,其显著优点为:1)通过连续流反应器替代传统的釜式反应器,液氯与3
‑
氯丙烯在反应装置内快速混合、快速反应,持液量较传统釜式要小的多,且传质传热效果好,反应安全可靠,生产风险低,同时减少液氯与3
‑
氯丙烯的混合时间,缩短反应时间;2)反应为低温、高压反应,无取代副产物生成,产品含量可达98.5%以上,收率达到
98.0%以上(以3
‑
氯丙烯计);3)连续流反应器内,物料不出系统,无气味产生、无有害气体逸出,且液氯较氯气通量大的多、用量精确可控,几乎不产生副产物,设备投入成本低,产能提升明显,是一种对环境友好、经济可行的工业化生产方案。
具体实施方式
11.为了使本发明的技术方案及优点更加清楚明白,以下结合实施例对本发明进一步详细说明,应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
12.实施例采用的连续反应装置为碳化硅微通道反应器,使用的是山东豪迈碳化硅材质微通道反应器,型号为rmcs1810,共5片,每片均可进料出料,反应器可耐压最大为1.8mpa,可耐温最高为200℃。
13.实施例1一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为
‑
10~0℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至
‑
10~0℃,通过流量计以2.5ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.6ml/min的流速进入微通道反应器内,通过控温系统,控制两者在
‑
10~0℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量431g,hplc检测含量98.5%,收率97.8%(以3
‑
氯丙烯计)。
14.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例2一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为0~10℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至0~10℃,通过流量计以2.7ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.7ml/min的流速进入微通道反应器内,通过控温系统,控制两者在0~10℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量432g,hplc检测含量98.9%,收率98.4%(以3
‑
氯丙烯计)。
15.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例3一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)和催化剂n,n
‑
二甲基甲酰胺6.9g混合均匀,事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为0~10℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至0~10℃,通过流量计以2.6ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.7ml/min的流速进入微通道反应器内,通过控温系统,控制两者在0~10℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件
下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量433g,hplc检测含量99.6%,收率99.3%(以3
‑
氯丙烯计)。
16.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例4一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)和催化剂乙腈5.6g混合均匀,事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为0~10℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至0~10℃,通过流量计以2.6ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.7ml/min的流速进入微通道反应器内,通过控温系统,控制两者在0~10℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量432g,hplc检测含量99.4%,收率98.9%(以3
‑
氯丙烯计)。
17.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例5一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)和催化剂n,n
‑
二甲基甲酰胺6.9g混合均匀,事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为
‑
20~
‑
10℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至
‑
20~
‑
10℃,通过流量计以2.5ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.8ml/min的流速进入微通道反应器内,通过控温系统,控制两者在
‑
20~
‑
10℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量434g,hplc检测含量98.6%,收率98.5%(以3
‑
氯丙烯计)。
18.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例6一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)和催化剂n,n
‑
二甲基甲酰胺6.9g混合均匀,事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为10~20℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至10~20℃,通过流量计以2.5ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.9ml/min的流速进入微通道反应器内,通过控温系统,控制两者在10~20℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量435g,hplc检测含量97.8%,收率98.0%(以3
‑
氯丙烯计)。
19.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).对比例1一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:
原料准备,在500ml四口烧瓶中,依次加入3
‑
氯丙烯100g(含量98%),催化剂n,n
‑
二甲基甲酰胺3g,通过氯气钢瓶以0.6g/min流速通入氯气,将反应瓶置于冷浴保持反应温度0~10℃,通氯至反应液密度增加,体系颜色变为绿色,共计通氯3h后停止通氯,继续保温0.5h取样中控,原料转化完毕经氮气赶气得到1,2,3
‑
三氯丙烷,重量187.2g,hplc检测含量97.5%,收率96.7%(以3
‑
氯丙烯计)。
20.对比例2一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:原料准备,在500ml四口烧瓶中,加入3
‑
氯丙烯100g(含量98%),通过氯气钢瓶以0.4g/min流速通入氯气,将反应瓶置于冷浴保持反应温度0~10℃,通氯至反应液密度增加,体系颜色变为绿色,共计通氯7h后停止通氯,继续保温0.5h取样中控,原料转化完毕经氮气赶气得到1,2,3
‑
三氯丙烷,重量186.5g,hplc检测含量94.6%,收率93.4%(以3
‑
氯丙烯计)。
21.对比例3一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:原料准备,在500ml四口烧瓶中,依次加入3
‑
氯丙烯100g(含量98%),通过氯气钢瓶以0.3g/min流速通入氯气,将反应瓶保持反应温度为20℃,通氯至反应液密度增加,体系颜色变为绿色,共计通氯11h后停止通氯,继续保温0.5h取样中控,原料转化完毕经氮气赶气得到1,2,3
‑
三氯丙烷,重量165.1g,hplc检测含量90.6%,1,1,2,3
‑
四氯丙烷8.5%,收率79.2%(以3
‑
氯丙烯计)。
22.对比例4一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯以20ml/min的流量通入到汽化器,将3
‑
氯丙烯汽化后,进入氯化塔,保护气氮气以20ml/min 的流量经过汽化器升温后通入到氯化塔,将氯气以9.5l/min的流量通入到氯化塔与3
‑
氯丙烯反应,维持塔内温度为50℃,连续进料1h,共计加入3
‑
氯丙烯1128g(含量98%),反应后得到的1,2,3
‑
三氯丙烷粗品,蒸馏后得到1,2,3
‑
三氯丙烷产品1951g,hplc检测含量84.5%,1,1,2,3
‑
四氯丙烷9.2%,收率77.4%(以3
‑
氯丙烯计)。
23.对比例5一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯以20ml/min的流量通入到汽化器,将3
‑
氯丙烯汽化后,进入氯化塔,保护气氮气以20ml/min 的流量经过汽化器升温后通入到氯化塔,将氯气以9.5l/min的流量通入到氯化塔与3
‑
氯丙烯反应,维持塔内温度为90℃,连续进料1h,共计加入3
‑
氯丙烯1128g(含量98%),反应后得到的1,2,3
‑
三氯丙烷粗品,蒸馏后得到1,2,3
‑
三氯丙烷产品1794g,hplc检测含量80.2%,1,1,2,3
‑
四氯丙烷10.4%,收率67.6%(以3
‑
氯丙烯计)。
24.通过以上实施例和对比例可以看出,利用连续流反应装置进行1,2,3
‑
三氯丙烷的制备,相较于对照例的普通釜式反应和气
‑
气反应,在收率、纯度等产品指标均有所提高,且反应为连续化反应,低温条件物料接触时间短,无取代副反应,设备投资成本低,对照气
‑
气反应在单位体积产能更高,使用连续流制备1,2,3
‑
三氯丙烷合成具有可行的工业化生产前景。
25.上述实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等
同替换方案为保护范围,即在此范围内的等同替换改进,也在本发明的保护范围之内。
‑
三氯丙烷的连续流制备方法
技术领域
1.本发明属于有机合成技术领域,具体涉及一种1,2,3
‑
三氯丙烷的连续流制备方法。
背景技术:
2.1,2,3
‑
三氯丙烷是重要的化工中间体,其cas号为96
‑
18
‑
4,分子式为c3h5cl3,其结构式为:。
3.1,2,3
‑
三氯丙烷为优良的溶剂,主要用作脱脂剂、去漆剂和电机洗涤用溶剂,还可用于生产三氯丙烯及农药矮壮素和燕麦敌等。其作为第二代烟碱类杀虫剂噻虫嗪、噻虫胺和医药利托那韦合成的关键起始原料,近几年来已逐渐引起了农药研究者们的广泛关注,1,2,3
‑
三氯丙烷产品经裂解反应可制备2,3
‑
二氯丙烯,通过与硫氰酸盐取代反应得2
‑
氯丙烯基异硫氰酸酯,再通氯气氯化可得到2
‑
氯
‑5‑
氯甲基噻唑中间体,该中间体随着烟碱类杀虫剂的快速发展,市场需求量巨大。
4.现有技术中,主要使用α
‑
氯丙烯(3
‑
氯丙烯)氯化法合成1,2,3
‑
三氯丙烷,该方法采用间歇釜式反应,最终通过精馏得成品,该方法虽然氯气的转化率可以达到95%,但纯度相对较低,且进料量难以控制。该反应方法生产成本高,反应温度控制难度大,并且产品质量不高,尤其收率较低,副产物多,污水废气处理难度大。
5.另外,de3432720a1提供了一种通过烯丙基氯与磺酰氯的反应制备1,2,3
‑
三氯丙烷的方法,具体为在液相中,在含氮碱、膦和/或氧化膦存在的条件下,在烯丙基氯中加入socl2,在约30℃至约70℃的温度范围内反应制备得到1,2,3
‑
三氯丙烷。该反应需要在无光的条件下进行,反应时间相对较长,反应中产生的so2、hcl气体逸出对环境造成污染,并且需要在减压条件下分离所得产物,甚至需要通过水洗来分离磺化副产物,不利于工业化生产。
6.cn108640811a提供了一种氯丙烯合成制备1,2,3
‑
三氯丙烷的方法,其采用气
‑
液反应的方式向氯丙烯中通入氯气、并通过冰水浴维持反应温度在20
‑
40℃下制备1,2,3
‑
三氯丙烯,但其反应时间较长、氯气消耗量极大且容易逸出,且需要经过碳酸钠水溶液清洗产品至中性并需要使用干燥剂及过滤,在经过复杂的后处理过程精制后的产品纯度至高为96.5%,并不十分令人满意。
7.cn108658722a提供了一种以3
‑
氯丙烯(α
‑
氯丙烯)和氯气为原料,采用气相合成法合成1,2,3
‑
三氯丙烷的方法,该反应需要先在超过45℃的高温下将3
‑
氯丙烯汽化,将汽化后的3
‑
氯丙烯和氮气或其他惰性气体与氯气通入到氯化塔中进行反应,得到的产物汽化后再经过蒸馏得到1,2,3
‑
三氯丙烷。虽然该方法反应为连续化反应,但反应所需大量氮气或其他惰性气体作为保护气体,投入成本高;反应所需氯气相对于液氯更易溢出,对生产人员的身体健康有一定的伤害;且为了保持气相条件,需要维持高温条件,温度过高产生一定的
副反应,副产物难以分离;气
‑
气反应通量小,单位产能受限,设备投入成本高且后期维护费用昂贵,对工人的综合素质要求高,从经济上考虑不适用于大规模工业化生产。
技术实现要素:
8.本发明的目的在于克服现有技术中的不足,提供了一种反应速度快,反应完全,成品含量高,产品收率高,生产产能高并且不存在三废,对环境友好的1,2,3
‑
三氯丙烷的连续流生产工艺。
9.本发明为了实现上述的技术目的,提供如下的技术方案:一种1,2,3
‑
三氯丙烷的连续流制备方法,通过柱塞泵同时将3
‑
氯丙烯和液氯泵入连续流反应装置内制备1,2,3
‑
三氯丙烷,具体反应方程式如下:;进一步地,所述的连续流反应装置为管式反应器;再进一步地,所述的管式反应器为微通道反应器;进一步地,进料方式为连续进料,用柱塞泵同时将3
‑
氯丙烯和液氯泵入;再进一步地,具体为两台柱塞泵同时将3
‑
氯丙烯和液氯泵入,制备得到1,2,3
‑
三氯丙烷;进一步地,所述3
‑
氯丙烯和液氯泵入前通过流量计;再进一步地,所述流量计为质量流量计或体积流量计;进一步地,反应温度为
‑
20~20℃;进一步地,反应温度为
‑
10~10℃;进一步地,反应温度为0~10℃;进一步地,3
‑
氯丙烯和液氯的摩尔比为1:1.0~1.5;进一步地,3
‑
氯丙烯和液氯的摩尔比为1:1.0~1.06;进一步地,催化剂为n,n
‑
二甲基甲酰胺或乙腈中的任一种;进一步地,在上述技术方案中,反应操作为:将原料3
‑
氯丙烯和液氯通过柱塞泵和流量计以定量的流速泵入微通道反应器,控制反应温度为
‑
20~20℃,反应结束后在0~10℃条件下接收反应液,得到1,2,3
‑
三氯丙烷;进一步地,在上述技术方案中,反应操作为:将原料3
‑
氯丙烯和液氯通过柱塞泵和流量计以定量的流速泵入微通道反应器,控制反应温度为
‑
10~10℃,反应结束后在0~10℃条件下接收反应液,得到1,2,3
‑
三氯丙烷;进一步地,在上述技术方案中,反应操作为:将原料3
‑
氯丙烯和液氯通过柱塞泵和流量计以定量的流速泵入微通道反应器,控制反应温度为0~10℃,反应结束后在0~10℃条件下接收反应液,得到1,2,3
‑
三氯丙烷。
10.由于采用了以上的技术,本发明与现有技术相比,其显著优点为:1)通过连续流反应器替代传统的釜式反应器,液氯与3
‑
氯丙烯在反应装置内快速混合、快速反应,持液量较传统釜式要小的多,且传质传热效果好,反应安全可靠,生产风险低,同时减少液氯与3
‑
氯丙烯的混合时间,缩短反应时间;2)反应为低温、高压反应,无取代副产物生成,产品含量可达98.5%以上,收率达到
98.0%以上(以3
‑
氯丙烯计);3)连续流反应器内,物料不出系统,无气味产生、无有害气体逸出,且液氯较氯气通量大的多、用量精确可控,几乎不产生副产物,设备投入成本低,产能提升明显,是一种对环境友好、经济可行的工业化生产方案。
具体实施方式
11.为了使本发明的技术方案及优点更加清楚明白,以下结合实施例对本发明进一步详细说明,应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
12.实施例采用的连续反应装置为碳化硅微通道反应器,使用的是山东豪迈碳化硅材质微通道反应器,型号为rmcs1810,共5片,每片均可进料出料,反应器可耐压最大为1.8mpa,可耐温最高为200℃。
13.实施例1一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为
‑
10~0℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至
‑
10~0℃,通过流量计以2.5ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.6ml/min的流速进入微通道反应器内,通过控温系统,控制两者在
‑
10~0℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量431g,hplc检测含量98.5%,收率97.8%(以3
‑
氯丙烯计)。
14.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例2一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为0~10℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至0~10℃,通过流量计以2.7ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.7ml/min的流速进入微通道反应器内,通过控温系统,控制两者在0~10℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量432g,hplc检测含量98.9%,收率98.4%(以3
‑
氯丙烯计)。
15.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例3一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)和催化剂n,n
‑
二甲基甲酰胺6.9g混合均匀,事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为0~10℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至0~10℃,通过流量计以2.6ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.7ml/min的流速进入微通道反应器内,通过控温系统,控制两者在0~10℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件
下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量433g,hplc检测含量99.6%,收率99.3%(以3
‑
氯丙烯计)。
16.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例4一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)和催化剂乙腈5.6g混合均匀,事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为0~10℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至0~10℃,通过流量计以2.6ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.7ml/min的流速进入微通道反应器内,通过控温系统,控制两者在0~10℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量432g,hplc检测含量99.4%,收率98.9%(以3
‑
氯丙烯计)。
17.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例5一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)和催化剂n,n
‑
二甲基甲酰胺6.9g混合均匀,事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为
‑
20~
‑
10℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至
‑
20~
‑
10℃,通过流量计以2.5ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.8ml/min的流速进入微通道反应器内,通过控温系统,控制两者在
‑
20~
‑
10℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量434g,hplc检测含量98.6%,收率98.5%(以3
‑
氯丙烯计)。
18.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).实施例6一种1,2,3
‑
三氯丙烷的连续流制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯230g(含量98%)和催化剂n,n
‑
二甲基甲酰胺6.9g混合均匀,事先置于含柱塞泵的原料瓶内,开启控温系统,使微通道反应器的温度为10~20℃,通过柱塞泵,将原料瓶中的3
‑
氯丙烯预冷至10~20℃,通过流量计以2.5ml/min的流速进入微通道反应器内,同时液氯通过流量计,以1.9ml/min的流速进入微通道反应器内,通过控温系统,控制两者在10~20℃内在微通道反应器内进行反应,反应停留时间为30s,于出口端在0~10℃条件下接收反应液,反应液经氮气吹扫后得到1,2,3
‑
三氯丙烷,重量435g,hplc检测含量97.8%,收率98.0%(以3
‑
氯丙烯计)。
19.产物核磁数据:1h nmr (dmso
‑
d6, 500 mhz) δ: 4.11
‑
4.14 (m, 1h), 3.62
‑
3.65 (q, 4h).对比例1一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:
原料准备,在500ml四口烧瓶中,依次加入3
‑
氯丙烯100g(含量98%),催化剂n,n
‑
二甲基甲酰胺3g,通过氯气钢瓶以0.6g/min流速通入氯气,将反应瓶置于冷浴保持反应温度0~10℃,通氯至反应液密度增加,体系颜色变为绿色,共计通氯3h后停止通氯,继续保温0.5h取样中控,原料转化完毕经氮气赶气得到1,2,3
‑
三氯丙烷,重量187.2g,hplc检测含量97.5%,收率96.7%(以3
‑
氯丙烯计)。
20.对比例2一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:原料准备,在500ml四口烧瓶中,加入3
‑
氯丙烯100g(含量98%),通过氯气钢瓶以0.4g/min流速通入氯气,将反应瓶置于冷浴保持反应温度0~10℃,通氯至反应液密度增加,体系颜色变为绿色,共计通氯7h后停止通氯,继续保温0.5h取样中控,原料转化完毕经氮气赶气得到1,2,3
‑
三氯丙烷,重量186.5g,hplc检测含量94.6%,收率93.4%(以3
‑
氯丙烯计)。
21.对比例3一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:原料准备,在500ml四口烧瓶中,依次加入3
‑
氯丙烯100g(含量98%),通过氯气钢瓶以0.3g/min流速通入氯气,将反应瓶保持反应温度为20℃,通氯至反应液密度增加,体系颜色变为绿色,共计通氯11h后停止通氯,继续保温0.5h取样中控,原料转化完毕经氮气赶气得到1,2,3
‑
三氯丙烷,重量165.1g,hplc检测含量90.6%,1,1,2,3
‑
四氯丙烷8.5%,收率79.2%(以3
‑
氯丙烯计)。
22.对比例4一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯以20ml/min的流量通入到汽化器,将3
‑
氯丙烯汽化后,进入氯化塔,保护气氮气以20ml/min 的流量经过汽化器升温后通入到氯化塔,将氯气以9.5l/min的流量通入到氯化塔与3
‑
氯丙烯反应,维持塔内温度为50℃,连续进料1h,共计加入3
‑
氯丙烯1128g(含量98%),反应后得到的1,2,3
‑
三氯丙烷粗品,蒸馏后得到1,2,3
‑
三氯丙烷产品1951g,hplc检测含量84.5%,1,1,2,3
‑
四氯丙烷9.2%,收率77.4%(以3
‑
氯丙烯计)。
23.对比例5一种1,2,3
‑
三氯丙烷的制备方法,具体步骤如下:原料准备,将3
‑
氯丙烯以20ml/min的流量通入到汽化器,将3
‑
氯丙烯汽化后,进入氯化塔,保护气氮气以20ml/min 的流量经过汽化器升温后通入到氯化塔,将氯气以9.5l/min的流量通入到氯化塔与3
‑
氯丙烯反应,维持塔内温度为90℃,连续进料1h,共计加入3
‑
氯丙烯1128g(含量98%),反应后得到的1,2,3
‑
三氯丙烷粗品,蒸馏后得到1,2,3
‑
三氯丙烷产品1794g,hplc检测含量80.2%,1,1,2,3
‑
四氯丙烷10.4%,收率67.6%(以3
‑
氯丙烯计)。
24.通过以上实施例和对比例可以看出,利用连续流反应装置进行1,2,3
‑
三氯丙烷的制备,相较于对照例的普通釜式反应和气
‑
气反应,在收率、纯度等产品指标均有所提高,且反应为连续化反应,低温条件物料接触时间短,无取代副反应,设备投资成本低,对照气
‑
气反应在单位体积产能更高,使用连续流制备1,2,3
‑
三氯丙烷合成具有可行的工业化生产前景。
25.上述实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等
同替换方案为保护范围,即在此范围内的等同替换改进,也在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。