作为多靶点激酶抑制剂的大环化合物的制作方法
- 国知局
- 2024-06-20 11:37:39
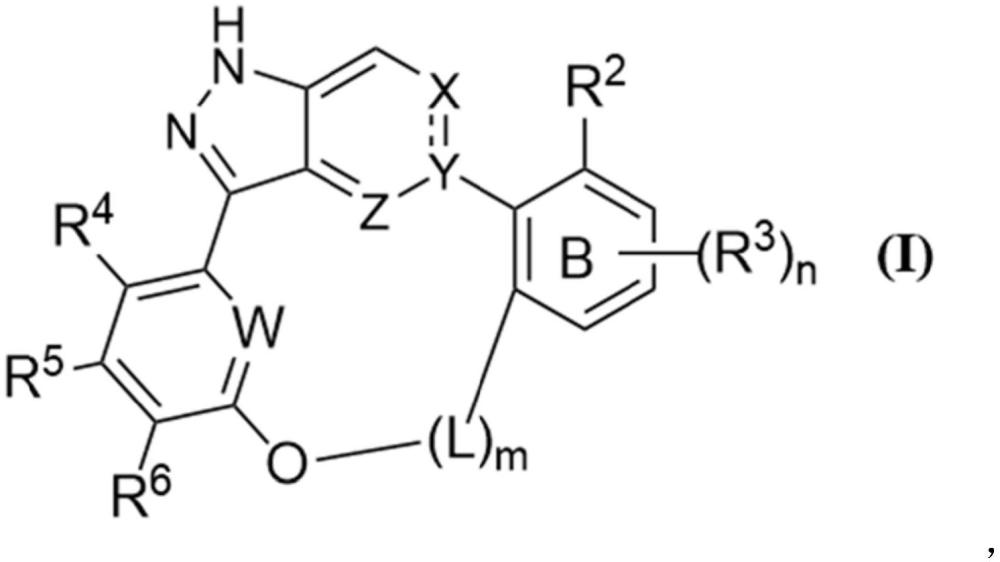
本发明涉及大环化合物,具体为一种具有多靶点激酶抑制活性的大环化合物及其药物组合物,还涉及此类化合物及其药物组合物在改善或治疗由蛋白激酶介导的疾病例如癌症的用途。
背景技术:
1、癌症,即恶性肿瘤,是威胁人类健康和生命的一类重大疾病,癌症的发病率和死亡率仍呈现明显上升趋势。肿瘤细胞的增殖、转移和凋亡与一系列信号传导通路的异常密切相关,肿瘤细胞的免疫逃逸和机体免疫功能的耗竭或受抑制也是癌症发生与发展的重要因素。因此,抑制肿瘤细胞信号通路中关键激酶的活性,同时通过激活免疫细胞,提高自身抗肿瘤的免疫力,可以有效抑制肿瘤细胞的生长与转移,治疗癌症病人,具有广阔的前景。
2、造血祖细胞激酶(hematopoietic progenitor kinase 1,hpk1)是一种丝氨酸/苏氨酸激酶,最初由造血祖细胞克隆得到(hu,m.c.等,genes dev.1996;10:2251-2264;keifer,f.等,the embo journal 1996;15:7013-7025),属于丝裂原活化蛋白激酶(mitogen-activated protein kinase kinase kinase kinase-4,map4k)家族。hpk1集中分布于淋巴器官或淋巴组织,如骨髓、淋巴结、胸腺等,且主要在免疫细胞(t细胞、b细胞、树突细胞)中表达(hu,m.c.等,genes dev.1996;10:2251-2264)。
3、研究表明,hpk1是t细胞受体(tcr)信号通路的负调节蛋白。tcr信号激活hpk1,激活的hpk1磷酸化slp-76的ser376残基,促进slp-76与14-3-3蛋白结合(di bartolo,v.等,j.exp.med.2007;204:681-691;shui,j.等,nature immuno.2007;8:84-91)。slp-76/14-3-3相互作用下调erk信号和钙离子流,并引发slp-76的泛素化和slp-76复合物降解,阻断tcr激活通路,从而抑制t细胞功能(lasserre,r.等,j.cell biol.2011;195:839-853)。
4、在体内实验中,hpk1敲除小鼠在抗原刺激下,t细胞功能增强,产生更多细胞因子,如il-2和ifn-γ(shui,j.等,nature immuno.2007;8:84-91;alzabin,s.等,j.immunol.2009;182:6187-6194;alzabin,s.等,cancer immunol.immunother.2010;59:419-429)。进一步研究表明,hpk1对免疫细胞的负调控依赖于其激酶活性。与野生型相比,阻断hpk1激酶活性小鼠的cd8+t细胞功能增强,可以更快的清除慢性淋巴细胞性脑膜炎病毒,以及更好地抑制肿瘤生长(hernandez,s.等,cell reports 2018;25:80-94)。在路易斯肺癌(llc)模型中,转染hpk1-/-t细胞的小鼠表现出比野生型更强的抗肿瘤免疫反应(sawasdikosol,s.等,immunol.res.2012;54:262-265)。研究揭示hpk1对b细胞(sauer,k.等,j.biol.chem.2001;276:45207-45216;tsuji,s.等,j.exp.med.2001;194:529-539;wang,x.等,j.biol.chem.2012;287:34091-34100;s.等,plos one,2010;5:e12468)、树突细胞(alzabin,s.等,j.immunol.2009;182:6187-6194)、nk细胞和treg细胞的免疫抑制作用源于也其激酶活性(liu,j.等,plos one,2019;14:e0212670)。
5、临床研究发现,与健康对照组相比,系统性红斑狼疮(zhang,q.等,j.autoimmun.,2011;37:180-189)和银屑病性关节炎(stoeckman,a.k.等,genes immun.2006;7:583-591;baltiwalla,f.m.等,mol.med.2005;11:21-29)病人的组织中hpk1水平显著下调,表明hpk1下调有助于增强自身免疫性反应。另一方面,在多种癌症中都观察到hpk1水平上调,例如急性粒细胞白血病(chen-deutsch,x.等,leuk.res.2012;36:884-888;chen-deutsch,x.等,cell cycle 2012;11:1364-1373),膀胱尿路上皮癌(wang.y等,mol.med.rep.2012;5:260-265),乳腺外佩吉特氏病(qian,y等,am j.dermatopathol.2011;33:681-686)和结肠癌(yang,h.s.等,mol.cell biol.2006;26:1297-1306)。
6、因此,hpk1是治疗肿瘤和病毒性疾病的很有希望的潜在靶标,开发hpk1激酶小分子抑制剂具有重要的临床前景。
7、fms样酪氨酸激酶3(fms-like tyrosine kinase 3,flt3)是一种受体酪氨酸激酶,在造血祖细胞和干细胞中表达,对造血细胞的存活、增殖和分化发挥重要作用。flt3的突变和异常表达导致白血病等血液疾病(gilliland,d.g.等,blood,2002;100:1532-1542;stirewalt,d.l.等,nat.rev.cancer,2003;3:650-665)。约30%成人急性髓性白血病(acute myel℃ytic leukemia,aml)患者体内存在flt3基因突变(nakao,m.s.等,leukemia,1996;10:1911-1918;kottaridis p.d.,blood,2001;98:1742-1759),是患者中最常见的基因突变和预后不良因素(abu-duhier f.m.等,british journal ofhaematology,2000;111:190-195;meshinchi s.等,clin.cancer res.,2009;15:4263-4269),使得flt3成为开发治疗此类肿瘤的小分子药物的重要靶点。
8、2017年,美国诺华公司开发的首个flt3抑制剂小分子药物midostaurin获批上市,用于联合治疗初次用药的flt3阳性aml患者。此后,由安斯泰来开发的gilteritinib,以及第一三共开发quizartinib先后上市,用于单药或联合治疗与flt3异常表达或突变有关的aml。尽管已有几款药物上市,但小分子flt3抑制剂作为aml的治疗方案仍存在较多问题,比如临床效果短暂,以及继发的耐药性。同时,临床数据表明flt3抑制剂药物能有效清除患者外周血原始细胞,但对骨髓效果较弱(bortheakur,g.等,haematologica,jan.2011;96:62-68)。以上问题的一个可能原因是存在替代的信号通路。因此仍需开发新一代flt3抑制剂,特别是能同时抑制多个信号通路的小分子化合物,用于未满足的临床需求。
9、血管内皮细胞生长因子受体(vascular endothelial growth factor receptor,vegfr),属于iii类受体酪氨酸激酶家族。其中vegfr2(kdr),也称激酶插入区受体(kinaseinsert domain receptor,kdr),广泛分布于血管内皮细胞中,在诱导肿瘤周围新生内皮细胞的生长、增殖和迁移过程中,起到非常重要的作用。
10、目前,已有多种vegfr2抑制剂获批,用于治疗相关肿瘤。2006年,美国辉瑞公司开发的vegfrs抑制剂小分子药物sunitinib获批上市,用于治疗胃肠道基质肿瘤和转移性肾细胞癌。作为一种多靶点抑制剂,sunitinib除能有效抑制vegfrs活性外,还对pdgfr、c-kit等多种激酶有抑制作用。多靶点的抑制作用使得药物不但有可能适用于多种适应症,而且可以降低因旁路信号激活引发的耐药性。此后有多款多靶点小分子抑制剂获批并取得了巨大成功,例如sorafenib、cabozantininb、lenvatininb等。
11、尽管目前已经公开了一些hpk1小分子抑制剂的专利申请,例如wo2018049152、wo2018049191、wo2018049200、wo2018049214、wo2018102366、wo2018183964、wo2019051199、wo2019090198、wo2019206049、wo2019238067、wo2020092528、wo2020103896、wo2021050964、wo2021213317、wo2022068848、wo2022089225、wo2022095904和wo2022253328等,但目前仍没有针对hpk1靶点的药物上市。特别的,同时抑制hpk1和其他激酶靶点的小分子化合物仍罕见报道,例如wo2020188467、wo2020235902和wo2023278483等。此类小分子化合物具有直接抑制肿瘤细胞生长同时激发自身免疫功能,从而消灭肿瘤细胞的作用。因此,开发上述这类具有高活性的新型小分子hpk1抑制剂是临床急需。
技术实现思路
1、本技术提供一种式(ⅰ)大环化合物,或其异构体、药学上可接受的盐、多晶型物、同位素标记物、活性代谢物或前药,
2、
3、其中,
4、w为n或ch;
5、x为n,c-cn或羰基,x和y之间的虚线表示该双键存在或不存在,其中:
6、当x为n或c-cn时,y为c,z为ch,x和y之间为双键;
7、当x为羰基时,y和z均为n,x和y之间为单键;
8、b环为苯环;
9、l在每次出现时各自独立地选自-o-、-ch2-、-ch(r1)-、-c(r1)2-、且与-o-相连的一个l不为-o-;且任意两个相邻的l不同时为-o-不同时为并且不同时为m个相互连接的l构成连接-o-与b环的连接基;
10、r1选自f和c1-3烷基;
11、r2选自h、f和c1-3烷氧基;
12、r3选自卤素、任选取代有1、2或3个r31a的c1-3烷基、任选取代有1、2或3个r31a的c3-6环烷基、c2-6炔基和氰基;
13、r4选自h、f和c1-3烷氧基;
14、r5选自:
15、1)h、f和c1-3烷氧基,
16、2)任选地取代有1或2个r51a的苯基和5-6元杂芳基,所述5-6元杂芳基包含1或2个选自n、o或s的成环杂原子,
17、3)任选地取代有1、2或3个r51b的5-7元单环脂杂环基或7-10元二环脂杂环基;
18、r6选自:
19、1)h、f和c1-3烷氧基,
20、2)下列结构中的一种,结构式中化学键末端的表示通过该键与式(i)结构上的其他原子相连:
21、
22、且r5和r6不同时为h、f或c1-3烷氧基;
23、条件是,当x为n,且(1)n为0,r4和r5均为h,且l各自独立地选自-o-或-ch2-时,或者,(2)n为0,r4和r5均为h,且-(l)m-为-(l)m-通过标记为“*”的键与b环相连结时,r6选自下列结构中的一种:
24、
25、r31a独立地选自f、羟基、c1-3烷氧基、氨基、c1-3烷氨基、和氰基;
26、r31b选自任选取代有1、2或3个r311的c1-3烷基和环丙基,其中r311选自f或羟基;
27、r51a独立地选自:
28、1)卤素、-ora1、-nra1rb1、-nra1c(=o)rb1、-nre1c(=o)nra1rb1、-nra1s(=o)2rd1、-nre1s(=o)2nra1rb1、-c(=o)ra1、-c(=o)ora1、-c(=o)nra1rb1和氰基,以及任选地取代有1、2或3个rg的c1-3烷基和环丙基,
29、2)任选地取代有1、2或3个r52的5-6元脂杂环基;
30、r51b独立地选自:
31、1)氧代基、f、羟基、c1-3烷氧基、环丙氧基、氨基、c1-3烷氨基、环丙氨基、c1-3烷基、环丙基和氰基,所述c1-3烷氧基、环丙氧基、c1-3烷氨基、c1-3烷基和环丙基任选地取代有1、2或3个rg;
32、2)任选地取代有1、2或3个rg的4-6元饱和脂杂环基;或者
33、3)连接于同一碳原子的两个r51b与所述碳原子一起形成
34、r52选自rg、c1-3羟基烷基和c1-3氰基烷基;
35、ra1、rb1和re1各自独立地选自:
36、1)氢;
37、2)任选地取代有1、2或3个rg的c1-3烷基和环丙基;
38、或者,
39、连接于同一氮原子的ra1和rb1与所述氮原子一起形成任选地取代有1或2个rg的3-6元脂杂环基;
40、rd1独立地选自任选地取代有1、2或3个rg的c1-3烷基和环丙基;
41、ra选自h,任选地取代有1、2或3个ra11的c1-3烷基,其中,ra11选自氟、羟基或氰基;
42、rb选自h、f、羟基、c1-3烷氧基、c1-3烷氨基、任选地取代有1、2或3个rb11的c1-3烷基、环丙基、氰基、其中,rb11选自氟或羟基;其中,中的成环碳原子任选地取代有1、2或3个c1-3烷基;所述中的成环碳原子任选地取代有1、2或3个rg;并且,连接于同一个碳原子上的两个rb不同时为h、羟基、c1-3烷氧基、c1-3烷氨基、
43、rg选自氧代基、f、羟基、c1-3烷氧基、c1-3氟代烷氧基、环丙氧基、c1-3烷基、c1-3氟代烷基、环丙基和氰基;
44、e1选自-ch(rx)-、其中rx选自
45、e2选自羰基、-ch2-、-c(ch3)2-、-ch(ry)-、其中ry选自异丙基、
46、e3选自-c(ch3)2-、-ch(ch2cn)-、
47、e4选自羰基、-ch2-、-c(ch3)2-、-ch(ch2cn)-、
48、e5选自-ch2-、-c(ch3)2-、-cf2-、其中rz选自h和c1-3烷基;
49、e环为3元饱和脂环或4~6元饱和脂杂环,所述饱和脂杂环包含1或2个选自n和o的成环杂原子,所述饱和脂杂环中成环碳原子任选地取代有1、2或3个c1-3烷基,饱和脂杂环中如果存在的成环n原子任选地取代有ra;
50、m为4、5或6;
51、n为0、1或2;
52、q为1或2。
53、本发明一实施方式还提供了一种药物组合物,包含上述的式(ⅰ)化合物或其药学上可接受的盐、水合物、溶剂化物、活性代谢物、多晶型物、同位素标记物、异构体或前药,以及药学上可接受的载体。
54、本发明一实施方式进一步提供了一种上述式(ⅰ)化合物或其药学上可接受的盐、水合物、溶剂化物、活性代谢物、多晶型物、同位素标记物、异构体或前药,或上述药物组合物在预防或治疗由蛋白激酶介导的疾病中的用途。
55、本发明一实施方式进一步提供了一种上述式(ⅰ)化合物或其药学上可接受的盐、水合物、溶剂化物、活性代谢物、多晶型物、同位素标记物、异构体或前药,或上述药物组合物在制备预防或治疗由蛋白激酶介导的疾病的药物中的用途,所述疾病包括肿瘤、骨髓增生异常综合征以及病毒引发的疾病中的一种或多种。
56、本发明一实施方式进一步提供了一种抑制hpk1激酶活性的方法,其包括向个体施用式(ⅰ)化合物或其药学上可接受的盐、水合物、溶剂化物、活性代谢物、多晶型物、同位素标记物、异构体或前药;
57、本发明一实施方式进一步提供了一种同时抑制多种蛋白激酶活性的方法,其包括向个体施用式(ⅰ)化合物或其药学上可接受的盐、水合物、溶剂化物、活性代谢物、多晶型物、同位素标记物、异构体或前药;所述蛋白激酶选自hpk1、flt3和kdr中的一种或多种。
58、本发明一实施方式进一步提供了一种治疗患者的由蛋白激酶介导的疾病或病症的方法,其包括向患者施用治疗有效量的式(ⅰ)化合物或其药学上可接受的盐、水合物、溶剂化物、活性代谢物、多晶型物、同位素标记物、异构体或前药。
59、本技术的化合物具有抑制多种蛋白激酶活性的作用,所述蛋白激酶包括hpk1、flt3和kdr等,能够用于治疗由这些蛋白激酶介导的疾病或病症。
本文地址:https://www.jishuxx.com/zhuanli/20240619/1780.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。