吡咯衍生物对碳同素异形体的加合物的制作方法
- 国知局
- 2024-07-27 12:12:59
根据treccani百科全书中的定义,在化学中的亲油描述了在油和脂肪中容易溶解的物质、原子、分子等。因此,在皂分子中,脂肪酸中的烃基残基是亲油的,而羧基是亲水-亲油的。
同样根据treccani百科全书中的定义,同素异形现象是在化学元素中,在固态中存在原子的不同排列和因此不同晶体结构相区分的形式。已知存在许多碳同素异形体。第一组是指碳原子的杂化。在金刚石中,碳原子是sp3杂化的。在其他的同素异形体中,碳原子是sp2杂化的。
在具有sp2杂化碳原子的同素异形体当中,可以提及下述:石墨烯、由若干层石墨烯层(从几个单位到数十个)组成的纳米石墨、石墨、富勒烯、纳米环、纳米锥、石墨烯纳米带、单壁或多壁碳纳米管,和炭黑(也称为灯黑)。石墨烯是一层碳原子并由此具有一个碳原子的厚度。单壁碳纳米管可以被视为卷起的石墨烯层。石墨、多壁碳纳米管和炭黑由石墨烯层构成。石墨由以约0.34nm的典型间距堆叠在结晶聚集体(aggregate)中的可变数量的石墨烯层组成。堆叠层的数量可以小于约10,但也可以达到几千。多壁碳纳米管可以被视为有若干层卷起的石墨烯层形成。正如已经提及的,单壁纳米管由一层形成,和多壁纳米管由若干层形成。在这些同素异形体的每个中,环作为基本单元存在。这些环典型地可以具有5或6个碳原子。在这些环中,存在在芳族多环体系上离域的在π轨道中的电子。这是可能的,因为环全部稠合并形成单一体系。芳族多缩合体系的最简单实例是芳族多环(包括:芘、菲、蒽)。具有sp2杂化碳的碳同素异形体构成具有不同平面度的芳族多缩合体系的等价物。为了使体系被定义为芳族,必须满足三个条件:(a)该体系必须是环状的,(b)环原子必须全部具有sp2杂化并且π电子的和必须满足休克尔规则(π=4n+2,其中n是包括0的整数),(c)该体系必须是平面的。在石墨烯的情况下,所有要求均满足。在非平面的碳同素异形体,例如富勒烯和碳纳米管的情况下,曲率影响平面性的状况。然而,这些体系仍可以被定义为芳族并它们代表例外。
其中碳是sp2杂化的碳同素异形体可以分为“纳米的”和“纳米结构的”。
当化学物质具有至少一个小于100nm的尺寸时,其被定义为“纳米的”。碳同素异形体如富勒烯、碳纳米管、石墨烯和纳米石墨是所谓的“纳米的”同素异形体。石墨烯是一层sp2杂化的碳原子,它具有一个碳原子的厚度并由此具有纳米尺寸。碳纳米管具有数纳米的直径。如上所述,由结晶聚集体形成石墨,而结晶聚集体本身由堆叠的石墨烯层形成。当堆叠的石墨烯层的数量低,从小于10到数十时,在与层正交的方向上结晶聚集体的尺寸范围为数nm到数十纳米。这些石墨因此被称为纳米石墨。单个管道可从它们互相缠结的混乱状态中分离。还可以将具有不同初始的堆叠层数的石墨进行剥离,得到具有低的堆叠层数的纳米石墨,以及石墨烯。
相反,超过一个世纪用于增强聚合物材料和用于许多其他应用的炭黑是“纳米结构的”。事实上它由具有纳米尺寸的初级颗粒组成,所述初级颗粒结合,以形成这些初级颗粒通过共价键保持在一起的聚集体。这些聚集体大于100nm。混合炭黑与聚合物基质的行为典型的以及使用所述基质典型的热机械应力不能使该聚集体分离成初级组分。聚集导致在初级颗粒之间产生孔隙,从而产生炭黑的特定的结构。孔隙的数量越大,该结构越大。这产生了纳米结构填料的定义。聚集体由此通过范德华力结合以产生附聚体(agglomerate),然而附聚体可以通过热机械应力被分离成初始聚集体。
其中碳是sp2杂化的碳同素异形体,如碳纳米管、石墨烯、石墨和炭黑,具有导电性和导热性。特别地,碳纳米管和石墨烯具有优异的机械和导电以及导热性能。它们具有纳米尺寸,即数纳米,在石墨烯的情况下是一个尺寸,和在碳纳米管的情况下是两个尺寸。纳米尺寸和特定的几何形状(在石墨烯的情况下,层状,和在纳米管的情况下,管状)给予它们大的表面积并由此能够与它们在其内掺入的基质建立大的界面面积,从而很大程度上影响其性能。由于它们的性能,碳同素异形体用于聚合物、塑料或弹性体基质,以及用于液体介质二者中,所述液体介质然后形成涂覆层。它们促进它们在其内掺入的材料的机械增强和导热性及导电性。
在基于sp2碳原子的碳同素异形体当中,迄今为止最常使用炭黑。考虑它在弹性体和热塑性基质中,在涂覆层中,和在调色剂中使用是足够的。炭黑提供机械增强,导电性,对紫外辐射的防护,以及最终产品的着色。
对“纳米”碳同素异形体,例如由若干层石墨烯层(从几个单位到数十个)组成的纳米石墨,和单壁或多壁碳纳米管与炭黑进行了许多研究。这两种碳同素异形体,和特别地能够具有工业应用。对石墨烯存在巨大的兴趣,但它的可获得性少使得它在工业上仍然是很少使用的产品。当使用“纳米”碳同素异形体,例如碳纳米管、石墨烯和纳米石墨时,对前述性能的改进尤其显著。而且,这些同素异形体在聚合物基质中具有明显的阻燃效果。
在聚合物基质的情况下,碳同素异形体可直接在所述基质内混合,通过常规的混合技术,形成最终产品,或者它们可形成预分散体的一部分,典型地碳同素异形体在预分散体中的浓度大于在最终产品中使用的浓度。类似地,在液体介质中的分散体的情况下,碳同素异形体可以形成待用于例如形成涂覆层的最终配方的一部分,或者可以存在于制备各种配方所使用的“母料分散体”中。
对于刚才提及的包含碳同素异形体的所有组合物,目的是获得同素异形体的最佳的分布和分散。在液体介质中的分散体的情况下,目的是首先获得分散体本身的稳定性,从而防止同素异形体的沉降。这些分散体的不稳定性导致在工业规模上缺少应用的开发。特别是对于“纳米”碳同素异形体来说,已经识别了这些问题。在聚合物复合材料的情况下,目的特别地是确保同素异形体与基质的最佳相互作用,和在材料的使用条件下的稳定相互作用。事实上,在含有碳同素异形体的聚合物复合材料的情况下可能遇到的最大问题是所述同素异形体与聚合物基质的相互作用不足。特别是对于“纳米”碳同素异形体来说,已经遇到到这一问题。这导致同素异形体的性能没有充分地转移到复合材料上并导致所述同素异形体本身的分散体不稳定,所述同素异形体倾向于形成聚集体,同时伴随最终材料性能的显著损害。
在制备含有sp2杂化碳同素异形体的复合材料中的主要目的因此是获得同素异形体的最佳分散。期望识别可获得所述最佳分散的标准。
bergin等人的文章,acsnano2009,第3(8)卷,第2340页给出了单壁碳纳米管在各种溶剂中的分散性。这些溶剂是:n-甲基-2-吡咯烷酮,1,3-二甲基-2-咪唑烷酮,n-乙烯基吡咯烷酮,n-二甲基乙酰胺,n-二甲基甲酰胺,1-环己基吡咯烷酮,n-丁基-2-吡咯烷酮,n-乙基-2-吡咯烷酮,1-苄基-2-吡咯酮,二甲基四氢-2-嘧啶酮,氧-吡咯烷丙腈,n-辛基吡咯烷酮,n-十二烷基吡咯烷酮和n-甲酰基哌啶。通过在离心之后,测定纳米管在分散体中的浓度,来测量纳米管的分散性。该文章给出了纳米管的分散性对它们在其内分散的介质的溶度参数的依赖性(作者报道了较好地依赖于由表面能数值推导出的溶度参数,而不是内聚能值)。描述这一依赖性的观点定位于具有不可忽视的斜率的直线。这表示基质溶度参数甚至小的变化可导致纳米管分散性的显著差别。该文章还报道了一些例外,从而表明需要进一步的研究。
然而,碳纳米管的溶度参数与它们在其内分散的介质的溶度参数之间的良好一致性仍然是获得碳纳米管良好分散性的优选参考点。事实上,在文章scientificreports(2014),4:7232中,使用了9种不同的聚合物基质。它们是:聚(苯乙烯-共-丁二烯)(在弹性体领域中称为sbr),聚(1,4-顺式-丁二烯),聚(1,4-顺式-异戊二烯),聚(丁二烯-共-丙烯腈)(在弹性体领域中称为nbr),聚(乙烯-共-1-丁烯-共-丙烯腈)(或氢化聚(丁二烯-共-丙烯腈))(在弹性体领域中称为氢化腈类橡胶),聚(环氧乙烷-共-表氯醇-烯丙基缩水甘油基醚),聚(乙烯-丙烯酸类单体)(在弹性体领域中称为丙烯酸类弹性体或acm),聚(偏氟乙烯-共-四氟乙烯-共-六氟丙烯)(在弹性体领域中称为氟橡胶)。在该项工作中,实验证明基质和纳米管的溶度参数之间的一致性对于允许在所述基质内形成纳米管的连续网络并由此获得导电性来说是重要的。事实上,列出了导电性与基质溶度参数相关性的曲线。据报道,采用氟橡胶作为基质,获得最好的分散。重要的是注意含有纳米管的复合材料显示出不同的导电性值,即使基质的溶度参数值变化很小。
因此,文献中的研究工作证明若我们希望实现碳纳米管在介质中或者在基质中的良好分散,重要的是纳米管和介质或基质的溶度参数类似。为了实现纳米管的最佳分散,需要具有基质特定的溶度参数值。非常令人感兴趣的观察结果是,为了获得碳纳米管在聚合物基质中的较好分散,而与所述基质的溶度参数无关,可以添加纳米管增容剂。仍然在文章scientificreports(2014),4:7232中,报道了添加小量氟橡胶作为纳米管的聚合物增容剂导致它们改进的分散和较高的导电性。
显然,碳同素异形体是亲油的。由此,和根据以上所述的内容,同样显然的是,原则上,可在亲油介质中实现碳同素异形体的良好分散。关于碳纳米管作为同素异形体,应当强调,碳纳米管由缠结在一起形成球的纳米纤维构成。当我们说到分散体时,我们意味着纳米管必须从球中解开并单独分散。为了实现解开,不仅仅是使用亲油介质的问题—需要优化分散亲油介质的性质。
鉴于它们的工业开发,获得碳同素异形体在亲油基质和尤其在亲油聚合物基质中的最佳分散是基本目的。事实上,迄今为止在工业规模上最重要的聚合物具有亲油性质。作为一个实例,我们可以提及:聚(乙烯),聚(丙烯),聚(苯乙烯),聚(丁烯),聚(异戊二烯),聚(丁二烯),聚(乙烯-共-丙烯),聚(异丁烯)。
在现有技术中,存在含有纳米尺寸的碳同素异形体的基于亲油基质的复合材料的许多实例。以下列出了含有“纳米”同素异形体的这种复合材料的一些实例。
在“carbonnanotube-polymerinteractionsinnanocomposites:areview,compositesscienceandtechnology72(2011)72-84”中描述了基于碳纳米管的复合材料。在“graphene-basedpolymernanocomposites.”polymer,52(1),5-25(2011)”中描述了基于石墨烯和纳米石墨的复合材料。在“multiwallcarbonnanotubeelastomericcomposites:areview”polymer,48(17),4907-4920(2007)中和在“theroleofcntsinpromotinghybridfillernetworkingandsynergismwithcarbonblackinthemechanicalbehavioroffilledpolyisoprene”macromol.mater.eng.,298,241-251(2012)中描述了碳纳米管在弹性体基质中的分散体。在“fillernetworkingofananographitewithahighshapeanisotropyandsynergismwithcarbonblackinpoly(1,4-cis-isoprene)-basednanocomposites”rubberchemistryandtechnology,第87卷,第2期,第197-218页(2014)中报道了在弹性体基质内纳米石墨的分散体。
然而,所有这些复合材料具有在单一成分颗粒水平下或者在单一纳米管或单一层状石墨烯或具有若干层石墨烯层和附聚体水平下分散的碳同素异形体。特别地,在“fillernetworkingofananographitewithahighshapeanisotropyandsynergismwithcarbonblackinpoly(1,4-cis-isoprene)-basednanocomposites”rubberchemistryandtechnology,第87卷,第2期,第197-218页(2014)中,示出了纳米石墨聚集体倾向于形成由在晶体结构内堆叠的若干石墨烯层形成的聚集体,当它们掺入到交联的弹性体复合材料内时。纳米尺寸的碳同素异形体在亲油基质中在组成颗粒水平(不管石墨烯层还是单一的纳米管)下分散的问题尚未解决,至少并未完全和以可控方式解决。
在现有技术中,列出了在亲油基质中分散“纳米”碳同素异形体的各种方法。
在“theroleofcntsinpromotinghybridfillernetworkingandsynergismwithcarbonblackinthemechanicalbehavioroffilledpolyisoprene”macromol.mater.eng.,298,241-251(2012)中,在密炼机内,利用混合的热机械能,在没有任何初步化学改性的情况下,混合纳米管与橡胶。这导致纳米管断裂,其显示出具有比混合之前的纳米管小的长度。
在文章carbon2012,50,4685-4695中,混合纳米管与在天然橡胶胶乳中的天然橡胶,用表面活性剂,例如十二烷基硫酸钠改性纳米管。然而,这一方法需要具有聚合物胶乳,并制备水性分散体,和因此是可能具有有限应用的方法。
在文章journalofappliedpolymerscience2012,125,e76-e84中,在溶剂,例如甲苯的辅助下,进行与天然橡胶的混合。除了使用非期望的芳族溶剂以外,显然的是使用溶剂复杂,和这意味着排除了任何工业开发。
而且,已知聚合物在溶剂中的溶解度取决于它们的分子和它们的立构规整度。已知低分子量的聚合物在高分子量的相同聚合物在其内不可溶的溶剂中变得可溶。而且,具有相同重复单元但具有不同立构规整度的聚合物在相同溶剂可能或者可能不可溶。实例是聚苯乙烯。无规立构聚苯乙烯在酮,例如甲乙酮中可溶,而全同立构聚苯乙烯和间同立构聚苯乙烯在甲乙酮中不可溶。全同立构聚丙烯在沸腾的庚烷中不可溶,而无规立构聚丙烯在沸腾的庚烷中可溶。因此,希望遵循使用溶剂在亲油基质中分散碳同素异形体的路径,我们可能考虑,原则上在能溶解低分子量聚合物和/或具有低的立构规整度的聚合物的溶剂中,分散碳同素异形体。尽管这些溶剂明显是有机分子,但它们还含有极性基团。典型实例是:醇,例如异丙醇和丁醇,酮,例如丙酮和甲乙酮,和酯,例如乙酸乙酯。从对健康和环境的影响角度考虑,例如相对于甲苯,这些物质不那么关键。通过与溶剂混合,可制备在聚合物基质内含有碳同素异形体的母料。这些母料然后可分散在高分子量聚合物的基质内。进行这一工序的条件是sp2杂化的碳同素异形体在所述溶剂内的良好分散体。
在文章compos.sci.technol.100(0)(2014)143和151中和在文章macromolecules2012,45,6045-6055中,起始点是用肼还原的氧化石墨烯。然后将其与天然橡胶胶乳混合和最后经历凝固。显然这一方法牵涉诸如将石墨氧化成氧化石墨的化学反应,一种采用强酸和强氧化剂,在苛刻条件下发生且具有一定危险程度的反应。这一反应导致碳原子杂化的强力改性。而且,它要求采用有毒成分,例如肼的还原反应。最后,正如已经提及的,它要求存在聚合物胶乳。
在文章compos.sci.technol.74(0)(2013)166和172中,首先使氧化石墨热膨胀,然后再次用肼还原,获得石墨烯或具有若干石墨烯层的聚集体。再者,该方法的复杂性是明显的。
现有技术具有非常丰富的关于炭黑在亲油聚合物基质中分散的数据与实验,正如在carbonblack,第2版,j.b.donnet,r.c.bansal,m.j.wang编辑,marceldekkerinc.,1993中已经报道的。在弹性体基质中使用大体积的炭黑。已知弹性体不可能具有实际应用,不仅若它们没有被硫化,而且若它们没有通过添加增强填料来增强的话。炭黑是超过一个世纪的用于增强弹性体的参考碳同素异形体。事实上,炭黑具有亚微米尺寸的组成颗粒且优选还具有所述颗粒的聚集体,在弹性体基质中不可溶,且具有显著高于基质本身的模量。因此,它可充当弹性体基质的增强剂。亚微米尺寸使得可能具有充足的表面积以供发挥增强作用。填料的表面积事实上转换成与聚合物基质的界面面积。界面面积事实上通过表面积与填料密度乘积及其体积分数给出。在增强填料和聚合物链之间强力的界面和良好的相互作用因此是机械增强的先决条件,因为它们允许将应力转移到能够储能的聚合物基质上。因此,明显地,在本文开头提及的“纳米填料”和“纳米”碳同素异形体具有大的潜力,考虑到它们的纳米尺寸和因此大的表面积以及大的界面面积。在含炭黑的弹性体基质的应用中,合适的是提及在轮胎胶料中的应用。借助热机械应力,进行炭黑与弹性体的混合。这一混合毫无疑问足以应用到轮胎胶料中。尽管如此,分散程度仍然不是最佳的,正如下文中讨论的。
事实上,在轮胎胶料的应用情况下,理想的状况是获得所需的机械增强和有限的能量耗散。例如在mark,j.e.,erman,b.,eirich,f.r.(eds.),thescienceandtechnologyofrubber,第3版,academicpress,sandiego,第367-400页,donnet,j.b.,custodero,e.,2005.,reinforcementofelastomersbyparticulatefillers中描述的增强理论教导了表面积造成在低应变下的机械增强。事实上,高表面积促进与聚合物链和与其他填料颗粒的强力相互作用,实质上基于非-键合力的相互作用,典型地范德华力。为了实现弹性体基质的有效增强作用,必须大量使用填料。典型地,astm标准胶料要求使用大于30份填料,每100份弹性体。在采用这一用量填料的情况下,填料超过其逾渗阈值,即它形成了网络。因此,不仅存在填料-聚合物相互作用,而且存在填料-填料相互作用。通过高表面积,促进这些相互作用。施加能量到含有增强填料的复合材料上和逐渐增加应变幅度,基于非键合相互作用的增强降低。因此根据称为佩恩效应的现象,粘弹模量降低。当弹性体复合材料经历高应变时,它遭受增强填料的增强作用。在高应变下的增强作用是由于聚合物基质与填料之间稳定的相互作用导致的,这种稳定的相互作用是由于填料的前述结构导致的,即在所述填料的初级颗粒之间的孔隙,它能接收聚合物链,锚定它们并将它们从粘稠的液体转变为增强填料。在轮胎中弹性体胶料的应用中,因此理想的是维持高应变典型的稳定增强并降低低应变下的增强,所述低应变通过施加能量到弹性体复合材料上而移除。也就是说,理想的是降低佩恩效应。事实上,这一能量被耗散,以克服非键合相互作用。对于在轮胎胶料中的应用来说,这反映在燃料消耗上以及增加的环境影响上。通过优化碳同素异形体的分散,降低佩恩效应,将其聚集体和/或基本粒子分开,并用一层弹性体覆盖它们。在弹性体胶料领域中,和尤其在轮胎领域中,这一目的尚未实现。目前,仅仅采用较高的混合能,可实现较好的分布与分散,但这导致聚合物基质降解。因此期望寻找改性碳同素异形体的方式,该方式允许碳同素异形体更好地分散在弹性体基质内。
在文献中没有报道过促进碳同素异形体在亲油基质中分散的碳同素异形体改性。
us2006/0045838描述了碳纳米管和选自以下的可溶聚合物之间的加合物:聚(噻吩)、聚(吡咯)、聚(芴)、聚(亚苯基)、聚(亚苯基亚乙炔基)、聚(亚苯基亚乙烯基)、聚(亚烷基芴)、聚(芴二噻吩)及其组合。改性剂的亲油性质促进在芳族溶剂,例如氯仿中分散。这些改性剂具有聚合物性质。因此,首先需要合成聚合物。因此必须通过在溶剂,例如氯仿中溶解,来生产与碳纳米管的加合物,从环境影响的角度来看,这也不是理想的。显然,这一方法不可应用到弹性体物品的工业生产上。
综述chem.soc.rev.,(2009),38,2214-2230报道了有机官能化(单壁)纳米管的方法。它们分为:(i)在官能化纳米管上形成酰胺和酯和(ii)加成反应。第一方法要求首先氧化纳米管并引入羧基。正如已经提及的,这一反应要求苛刻的条件,和强酸与强氧化剂,且具有一定程度的危险性。难以想象它的大规模工业开发。而且,氧化必须紧跟着进一步的化学反应。这一方法因此在合适地配备的化学实验室中似乎是可行的。当然它不可能在加工设备中就地进行。加成反应(第二种方法)必须用元素氟来氟化,通过用十八烷基胺官能化,接着添加苯基(溴代二氯甲基)汞作为二氯卡宾源来加成卡宾,使用烷基叠氮甲酸酯,加成氮宾,1,3-偶极环加成,亲核加成(所述亲核加成例如使用溴代丙二酸二乙酯和1,8-二氮杂双环[5.4.0]十一碳烯,导致环丙烷化),使用自由基的直接加成,和使用重氮盐的直接芳化。所有这些方法需要使用化学反应,有时在官能化纳米管上进行的化学反应。待使用的设备和化学反应的典型条件是明显的。对于制备小样品来说,它们似乎是典型的方法。从对健康的影响角度考虑,有时需要使用关键的化学品。最后,不可能想象在拟用于混合的机器内就地进行这些反应。在这一综述中,使用通过狄尔斯-阿德耳反应,与碳纳米管反应的改性剂是让人泄气的,牢记导致加合物破坏的可逆的狄尔斯-阿德耳反应仅仅通过加热加合物本身可容易地发生。
重要的是注意为改性碳同素异形体而选择的每一路线的特征在于某一类型的“改性化学”,其牵涉某一组的官能团。
期望能制备具有sp2杂化碳原子的碳同素异形体在液体介质和亲油性质的聚合物基质二者中,特别地在亲油性质的聚合物基质中的稳定分散体。
期望在吡咯衍生物和sp2杂化碳同素异形体之间的分散体供我们使用,其中通过改性碳同素异形体,使之保留它们的sp2性质基本上不变来获得所述分散体。
而且,期望这一改性以简单的方式发生,和通过将试剂进料到同素异形体本身上并原位,也就是说在聚合物基质本身内进行改性反应,从而进行改性反应。
还期望能使用简单的技术和采用降低的环境影响以供进行改性反应。在聚合物基质内的分散体情况下,期望避免使用任何溶剂。
因此期望通过进行非键合或超分子相互作用,或者通过形成共价键,但与此同时没有显著改性已经改性的同素异形体的电学性质,采用含有能与碳同素异形体中的芳环相互作用的官能团的化合物,进行碳同素异形体的改性。
为了改性碳同素异形体,期望能使用含有促进与碳同素异形体相互作用的官能团,且还含有促进与聚合物基质相互作用的另一或其他官能团的化合物。
期望与碳同素异形体形成加合物的化合物族,所述的化合物族特征在于仅仅一类促进与碳同素异形体相互作用的官能团,因此可进行该化合物和同素异形体之间可再现的相互作用,以及特征在于促进与液体介质或聚合物基质相互作用的各类官能团,进而使得也可以在液体和亲油性质的基质中制备稳定分散体,从而优化与亲油基质的相互作用。
还期望在促进与液体介质或聚合物基质的碳同素异形体有利地相互作用的官能团当中,还存在能促进与溶剂相互作用的官能团,所述溶剂由有机分子组成,但还含有极性基团,以便能利用非常大数量的溶剂以及用于延伸碳同素异形体增容作用的可能性目的。
还期望改性碳同素异形体的非常多样的方法,例如使得形成改性同素异形体的化合物的试剂成为可能,其中所述化合物被进料到同素异形体本身上。因此,期望能进行改性剂的合成,和当然随后在同素异形体上改性这二者。
最后期望由同素异形体和改性剂形成的加合物应当使得可获得碳同素异形体的稳定分散体,以便随着时间流逝,维持它们的特性。
本发明的一个目的因此是,通过引入至少一个亲油取代基,改进碳同素异形体在亲油介质中的分散性,提供其中碳是sp2杂化的碳同素异形体的改性方法。
本发明的一个目的因此是,提供其中碳是sp2杂化的碳同素异形体与至少一种含有能与碳同素异形体的芳环相互作用的官能团以及改进同素异形体在亲油介质和基质中分散性的官能团的化合物之间的稳定加合物。
本发明的一个目的是在碳同素异形体和以上所述的化合物之间形成稳定的加合物,且没有显著改变同素异形体中碳原子的杂化和进而没有显著改变它们的性能。
本发明的一个目的因此是,由简单可容易获得的化学品为起始,提供其中碳是sp2杂化的碳同素异形体之间的稳定加合物。
本发明的一个目的是为了形成加合物,使用下述化合物族,该化合物族使用与形成加合物相同的实验条件,且与此同时可调节改性同素异形体的化学性质。
本发明的一个目的是根据绿色化学原理,因此使用具有降低的环境影响的技术,以简单和可再现的方式,采用简单的实验条件,进行该化合物与碳同素异形体之间的反应。
本发明的一个重要目的是进行形成下述化合物的反应,所述化合物在同素异形体本身上与碳同素异形体形成加合物,进而省去了该工艺的一个基本步骤,即合成并分离该化合物的步骤。
借助式(i)的化合物与含有sp2杂化碳原子的碳同素异形体,实现本发明的这些和其他目的
其中r1,r2,r3,r4独立地选自:氢、c1-c3烷基、c2-c18直链或支链烯基或炔基、芳基、c1-c18直链或支链烷基-芳基、c2-c18直链或支链烯基-芳基、c2-c18直链或支链炔基-芳基、杂芳基,
和x选自:
其中r5和r6独立地选自:氢、c1-c18烷基、c2-c18直链或支链烯基或炔基、芳基、c1-c22直链或支链烷基-芳基、c2-c22直链或支链烯基-芳基、c2-c22直链或支链炔基-芳基、杂芳基,
或者r5或r6每一个或二者是
其中m=0、1、2和n=1-30
其中若r5或r6仅仅之一是
其中m=0、1、2和n=1-30
则另一个选自:氢、c1-c18烷基、c2-c18直链或支链烯基或炔基;
或者r5和/或r6是:
其中n=0、1、2、3
和r7,r7′,r7"独立地选自:c1-c4烷基;c1-c4氧-烷基
或r5和/或r6是:
其中n=0、1、2、3
和r8选自:c1-c4烷基;
或者r5和/或r6是:
其中n是1至10的整数;
r9选自:氢、烷基、芳基、苄基、胺、烷基胺、芳基胺、苄基胺、氨基芳基;
r10,r11,r12,r13和r14独立地选自:氢、c1-c18烷基、c2-c18直链或支链烯基或炔基、1-(4-氨基环己基)亚甲基。
按照这一方式,获得含有具有sp2杂化碳原子的碳同素异形体的加合物,它在许多亲油性质的基质中可分散,且可在其中需要维持所述同素异形体的特性的方法中使用。
优选地,所述r1,r2,r3,和r4独立地选自:h,ch3,ch2ch3,苯基。
优选地,碳同素异形体或其衍生物选自:炭黑,富勒烯,单壁或多壁碳纳米管,石墨烯,具有2至10000石墨烯层数量的石墨。
优选地,所述碳同素异形体衍生物含有选自下述中的官能团:
-含氧官能团,优选羟基、环氧基;
-含羰基的官能团,优选醛基、酮基、羧酸基;
-含氮原子的官能团,优选胺基、酰胺基、腈基、重氮盐、亚胺基;
-含硫原子的官能团,优选硫基、二硫基、硫醇基、砜基、亚磺酸基和磺酸基。
优选地,碳同素异形体衍生物是氧化石墨或氧化石墨烯。
因此,我们可以根据我们的处置,使用大范围的含有sp2杂化碳原子的碳同素异形体。
本发明进一步的目的是提供制备根据前述一项或多项权利要求的加合物的方法,该方法包括下述步骤:
i.在质子或非质子极性溶剂中提供式(i)化合物的溶液;
ii.在制备步骤i.中提到的溶液所使用的质子或非质子极性溶剂中提供碳同素异形体的悬浮液;
iii.混合所述溶液和所述悬浮液;
iv.从步骤iii.中获得的混合物中移除所述溶剂;
v.向所得混合物提供热能和/或机械能和/或光子辐射能。
优选地,在50℃至180℃的温度下提供热能15至360分钟。
优选地,提供机械能15至360分钟的时间。
优选地,在200至380nm的波长下提供光子辐射能30至180分钟的时间。
以下描述了以合成起始单体为开始,本发明加合物的制备方法。
根据本发明,由下述通式的伯胺为起始,通过合成式(i)的化合物,获得所描述的组合物:
r-nh2
其中r选自:氢,c1-c3烷基,直链或支链c2-c22烯基或炔基,芳基,直链或支链c1-c22烷基-芳基,直链或支链c2-c22烯基-芳基,直链或支链c2-c22炔基-芳基,杂芳基。
这些伯胺大部分可商购或者可以根据制备伯胺的经典技术制备[twgrahamsolomons,craigbfryhle;chimicaorganica,2008,zanichelli]。
为了获得式(i)的化合物,使伯胺与下述通式的二酮反应:
其中r1,r2,r3,r4独立地选自:氢,c1-c22烷基,直链或支链c2-c22烯基或炔基,芳基,c1-c22烷基-芳基,直链或支链c2-c22烯基-芳基,c2-c22炔基-芳基,杂芳基。
作为一个实例,下文描述了通过两步法,制备式(i)的吡咯衍生物,亦即1-己基-2,5-二甲基-1h-吡咯。
在不存在溶剂和没有添加催化剂的情况下,在温度高于100℃,优选150℃下,使1-己胺与2,5-己二酮在等摩尔量下反应等于约5小时的时间,高产率地获得1-己基-2,5-二甲基-1h-吡咯,产率等于约95%。
根据本发明的碳同素异形体是或多或少反应性的芳族体系,和因此受到各类分子间的相互作用。
这些包括堆叠。事实上,堆叠定义为芳族分子的堆叠排列。事实上,包含芳族环的分子倾向于自发地在彼此上堆叠排列。这产生芳族相互作用(或π-π相互作用)的根源,其意味着在包含芳族基团的有机化合物之间建立的非共价类型的键,考虑到在π-共轭体系中p轨道的分子间重叠。该类型的相互作用使得该键更加稳定,因为其增加了π电子的数量。
通过各类化学反应,例如在综述chem.soc.rev.,(2009),38,2214-2230中列出的那些化学反应,碳同素异形体也可导致共价键的形成。
根据本发明,生产下述组合物,其中在碳同素异形体和在氮原子上带有亲油取代基的吡咯衍生物之间存在稳定的相互作用。5元杂环,例如吡咯被称为“富电子”,因为该芳环由5个原子和6个π电子组成。每一原子的电子密度因此大于苯环。这一五边形结构和存在杂原子使得该体系不那么稳定,因此,反应性远比苯大得多。已知在下述三个富电子的杂环当中,相对于呋喃和噻吩,吡咯的反应性在中间。这些富电子的杂环的反应性不如二烯烃,但反应性大于苯。事实上,它们能得到富电子二烯烃典型的反应。
根据本发明,术语加合物是指通过可导致形成如下才者的的相互作用/反应获得的化合物,超分子化合物,即其中起始化学个体通过非键合的相互作用来相互作用,以及含有共价键的化合物。
在导致加合物形成的反应中,可以假设以下两种类型的相互作用/反应:
(i)π-π相互作用。这一形式的相互作用可以存在于具有π电子,和因此具有sp2或sp杂化的体系之间。该相互作用在一对π电子和一个σ轨道之间,或者在一个σ轨道和一个π轨道的电子之间,或者在两个π轨道的电子之间。该类型的加合物也被称为“π配合物”。如上所述,该类型的反应导致堆叠。
(ii)导致在吡咯环和碳同素异形体之间形成共价键的反应。典型的反应是环加成反应,例如狄尔斯-阿德耳反应,它可在石墨化合物的芳环与含有吡咯环的化合物或由其衍生的含有吡咯环的化合物之间发生。这一最后提及的衍生物可以在与碳同素异形体相互作用的条件下,特别是温度和时间下形成。
同素异形体本身可以在吡咯化合物和石墨碳同素异形体之间的相互作用/反应类型方面起到重要作用。事实上,石墨,纳米石墨和衍生物,例如氧化石墨和氧化石墨烯的催化作用是已知的。在描述这一催化行为的科学文献中的文章例如是navalon,sergio等人的“carbocatalysisbygraphene-basedmaterials.”chemicalreviews,114.12(2014):6179-6212。
如上所述,碳同素异形体,特别是“纳米”同素异形体如富勒烯、碳纳米管、石墨烯和纳米石墨可以包含不同种类的官能团。根据本发明,在具有吡咯环的分子之间进行加成反应,该吡咯环在氮原子上带有亲油取代基。可通过在亲油取代基上存在的官能团和在碳同素异形体上存在的官能团发生加合物的形成。
根据本发明,通过在碳同素异形体和在氮原子上带亲油取代基的吡咯化合物之间的加成反应形成的加合物可以是可逆的。加合物的可逆性可以是不同反应参数如温度、时间、溶剂使用的函数。
现描述含式(i)化合物的加合物的制备方法。
在本发明可能的一种实施方案中,该方法包括下述步骤:
a)制备至少一种式(i)化合物在溶剂中的溶液,所述溶剂可以是:(i)选自醇,羰基溶剂如丙酮,酯例如乙酸乙酯,二甲亚砜,乙腈,醚中的质子或非质子极性溶剂,(ii)非质子溶剂,例如戊烷,己烷,庚烷,及其高级同系物和异构体;
b)在制备式(i)化合物的溶液所使用的极性或非极性溶剂中制备碳同素异形体的悬浮液;
c)借助机械或磁搅拌体系,或者例如借助超声浴,通过用超声设备的超声处理,混合式(i)化合物的溶液和碳同素异形体的悬浮液;
d)从所得混合物中移除溶剂;
e)任选地,向所得混合物提供热能和/或机械能和/或光子辐射能。
在步骤a)-c)中描述的工序导致获得纳米填料和在氮原子上带亲油取代基的至少一种吡咯化合物的均匀分散体,和然后获得在氮原子上带亲油取代基的吡咯化合物在碳填料上的均匀分散体。在能量转移到碳纳米填料和氮原子上带亲油取代基的至少一种吡咯化合物之间的加合物上的下一步之前,移除溶剂。
术语溶剂涉及氮原子上带亲油取代基的吡咯化合物且显然不涉及碳同素异形体,对于碳同素异形体来说,溶剂仅仅充当分散介质。优选地溶剂应当环境友好的。
在本发明说明书的其余部分中,术语“碳同素异形体”和“碳填料”可互换使用。
一般地,由于碳的化学性质导致相当难以在液体基质内分散碳填料。使用超声使得可在合理的时间内进行分散,并改进碳填料的分散的均匀性(甚至仅仅数秒)。而且,使用超声处理可在变化的程度上将碳纳米填料分离成基本单元。碳纳米管可以从它们与其他管道互相缠结的混乱状态中分离成单个管道。使用低功率超声发生器,例如经典超声浴是可取的。采用合适的溶剂还可以进行具有不同起始堆叠层数的石墨的至少部分剥离。具有低堆叠层数的石墨具有纳米尺寸并被称为纳米石墨。因此,优选纳米填料预先与液体接触,以通过超声处理和取决于纳米填料,获得或者所谓解开的碳纳米管,或者石墨或纳米石墨的或大或小程度的剥离。这一工序导致纳米填料和氮原子上带亲油取代基的吡咯化合物之间改进的接触,且还导致纳米填料暴露面积的增大。
根据本发明,术语“声化学”是指研究在用超声辐照的溶液中发生的化学反应的物理化学学科。对于在给定阈值以上的范围的场强度而言,该辐照导致在溶液中的空化现象。存在于溶液中的气体微腔(气泡)经受通过震荡声压场诱导的后续的膨胀和收缩,尺寸增加并随后内爆,产生非常高的温度和压力的区域。在这些极限条件中,在有机物质的合成、聚合方法以及有毒有害物质的降解领域中相当大兴趣的化学反应可以发生。在应用超声处理技术的情况下,还可以得到无定形材料,所述材料在超声处理的典型极限条件之外将自然倾向于结晶。
可通过例如真空蒸发,喷雾干燥等移除溶剂的任何合适的方法,发生根据步骤d)从所得混合物中移除溶剂的方法。
在从含有式(i)化合物和碳同素异形体的混合物中移除溶剂之后获得的混合物可经历进一步的步骤e),其中将能量转移到组合物中。
在能量向由含键合至二醇的吡咯环的分子和碳同素异形体组成的体系转移的情况下,进行导致加合物形成的加成反应。进行能量转移以便改进含键合至二醇的吡咯环的分子和碳同素异形体之间的相互作用。在不存在能量转移的情况下,在键合至二醇的吡咯环和碳同素异形体之间的相互作用较弱。较弱的相互作用可引起碳同素异形体从含键合至二醇的吡咯环的分子中部分释放,特别是如果加合物处于极性性质的环境中。
可以被转移到组合物以允许其形成的能量形式是:
-机械能
-热能
-光子
机械能
通过机械方法,处理通过以上在步骤a-c中描述的方法获得的在纳米填料和氮原子上带亲油取代基的至少一种吡咯化合物之间得到的混合物。
机械处理由将所得粉末(纳米填料/sp)置于配备有不锈钢球的罐中组成。一旦密闭,则将罐置于行星式混合机中并使其以200至500rpm的速度旋转1至360分钟的时间。随后将粉末立即倒出。
所提及的机械处理用于诱导无序化(在石墨的情况下剥离)以得到在纳米填料上sp更好的分布,并且用于诱导显著更加稳定的相互作用的形成这二者。
这是可能的,因为通过使干混合物经受机械力诱导在干混合物上的化学反应的可能性是化学领域中已知的。虽然机械化学是化学领域的分支,其不是非常熟知,但是其由于其环境友好的特性而引起了巨大的兴趣。机械化学方法可以通过简单地使用研钵和研杵或使用操作简单的比较庞大的系统,如在制药和食品工业中使用的球磨机触发。
所谓的行星式球磨机具有在旋转平台上保持在垂直位置的圆柱形反应器、罐。在带有包含球的罐的(球)磨机中,利用典型地数量为5至50个球之间的碰撞。与给定机械化学转换有关的给定磨操作的效率与球和罐内壁之间的碰撞频率和转移的机械能密切相关。这些量转而取决于球的动力学,它们的数量和尺寸,(球)磨机的震荡或工作频率以及在反应器内部的粉末总量。
热能
通过热方法,处理通过在以上步骤a-c中描述的方法获得的在纳米填料和氮原子上带亲油取代基的至少一种吡咯化合物之间得到的混合物。
热处理由将所得粉末(纳米填料/sp)置于带有冷凝器的反应烧瓶或密封小瓶中组成。一旦将反应器置于热板上,在130至180℃的温度下进行反应。保持加热2小时的最低值至12小时。加热处理诱导稳定的相互作用的形成。
光子
使用具有合适波长的灯,通过辐照方法,处理通过在以上步骤a-c中描述的方法获得的在纳米填料和氮原子上带亲油取代基的至少一种吡咯化合物之间得到的混合物。
光子处理由将所得粉末(纳米填料/sp)置于实验室结晶器中形成薄层或者将该粉末置于密封的石英小瓶中组成。一旦将反应器置于配备有254nm低压汞灯的暗室中(或者使用配备有相同类型的灯的反应器),将混合物辐照30至180分钟的时间变量。在该时间之后,将混合物倒出并分析。
采用根据本发明的加合物可以得到碳纳米填料在水性介质和其他基底(如聚合物共混物或橡胶)二者中稳定的悬浮液,以便得到均匀的产物,所述均匀的产物具有碳纳米填料的特定的性质,如高机械性能、高导电性、耐高温性、阻燃性能。
采用根据本发明的加合物还可以得到炭黑填料在各种基底上均匀、连续的层,以便得到高度导电的表面。
以下描述了根据本发明的包含氮原子上带亲油取代基的吡咯化合物和碳同素异形体的加合物的制备的一些实施例。
通过以下列出的实施例将更好地阐述根据本发明的加合物,所述实施例阐述了所述加合物制备方法的步骤。
由例如在附图中阐述的优选实施方案的说明,将更好地理解本发明的特征和优点,其中:
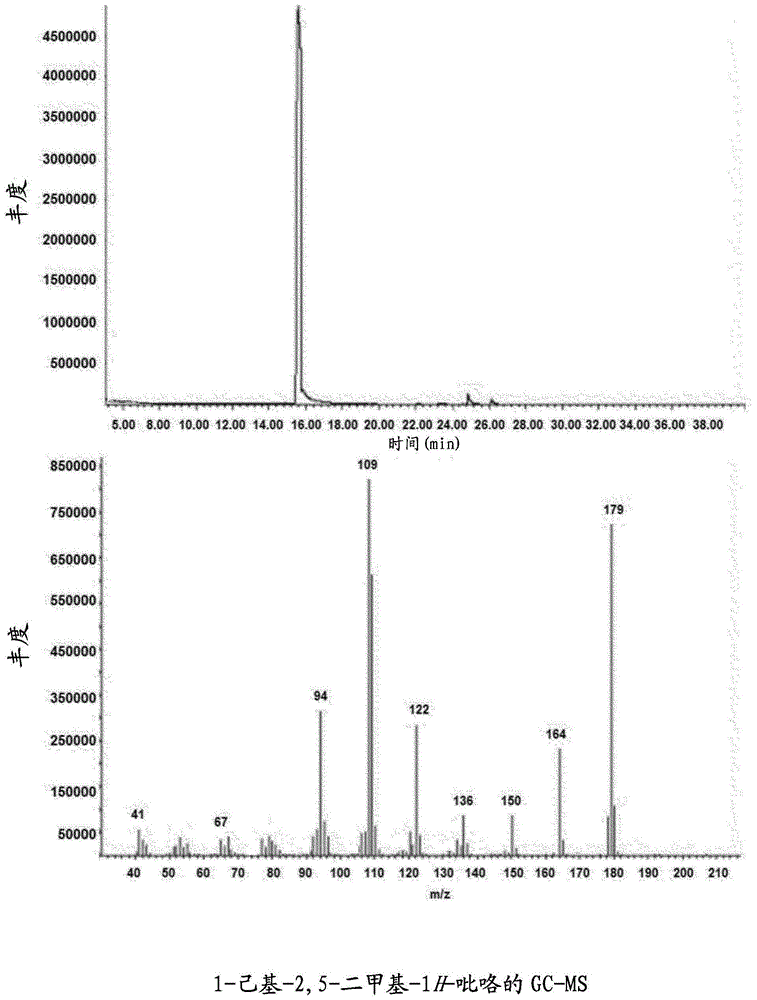
-图1示出了在gc-ms分析1-己基-2,5-二甲基-1h-吡咯之后获得的色谱图和质谱;
-图2示出了在cdcl3中的1-己基-2,5-二甲基-1h-吡咯在400mhz下的1hnmr光谱;
-图3示出了1-己基-2,5-二甲基-1h-吡咯与cbn326的加合物的ftir光谱;
-图4示出了在gc-ms分析1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷之后获得的色谱图和质谱;
-图5示出了1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷与纳米石墨的加合物的ftir光谱;
-图6示出了在cdcl3中的2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯在400mhz下的1hnmr光谱;
-图7示出了2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯与纳米石墨的加合物的ftir光谱;
-图8示出了在cdcl3中的3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺在400mhz下的1hnmr光谱;
-图9示出了在cdcl3中的o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇在400mhz下的1hnmr光谱。
实施例
在下述实施例给出的合成中所使用的所有化学品获自aldrich且在没有进一步纯化的情况下使用。
在以下列出的实施例中获得的组合物分析如下:
-通过红外光谱法的分析(使用kbr压片的ft-ir):使用重量比为1:500的加合物/kbr和约80mg的混合物以形成压片。通过傅里叶变换ir分光光度计(具有atr选项的varian640-irft-ir光谱仪)分析该压片。在2.5至20μm(或4000至500cm-1)的范围内辐照样品。
-uv光谱法:使用pasteur移液管,将加合物悬浮液(3ml)置于具有1cm光路长度的石英比色皿(体积1或3ml)中并使用uv-vis分光光度计分析。该仪器用纯溶剂重置并记录200-340nm的uv光谱。记录所用溶剂的空白样。该uv-可见光谱示出吸收强度作为200至750nm的辐射波长的函数。
-在溶剂中的稳定性:在处理之后,将粉末置于实验室小瓶中,加入合适的溶剂(浓度为1mg/ml)并将其超声处理10分钟,在超声处理最后(在时间t=0处),通过uv光谱分析。然后在1天和1周之后重复该分析。
实施例1-合成1-己基-2,5-二甲基-1h-吡咯(己基吡咯,hp)
经验式:c12h21n
摩尔质量:179.30
向配备有磁搅拌器的100-ml单颈烧瓶中引入3.75g(0.151mol)己胺和4.25g(0.151mol)的2,5-己二酮。在150℃下搅拌该混合物5小时。
以浅黄色油状物形式分离该产物,产率为95%并通过与质谱相连的气相色谱(gc-ms)和核磁共振(nmr)分析。图1示出了与质谱相连的色谱图。在这一实验中测定的质量对应于化合物1-己基-2,5-二甲基-1h-吡咯的理论质量。图2示出了1h-nmr光谱。
实施例2-采用在作为碳同素异形体的石墨上负载的反应物合成1-己基-2,5-二甲基-1h-吡咯
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向配备有磁搅拌器的100-ml单颈烧瓶中引入3.75g(0.151mol)己胺,4.25g(0.151mol)的2,5-己二酮和10g石墨。该烧瓶配备有磁搅拌器并使混合物在150℃的温度下进行缓慢旋转2小时。然后使该反应混合物达到室温。取出固体样品并置于试管中。在室温下添加氘代氯仿(cdcl3)。室温下手动搅拌试管2分钟。倾析试管内包含的悬浮液。通过1h-nmr光谱分析这一液体,其揭示与图2中所示相同的峰,并因此证实了化合物1-己基-2,5-二甲基-1h-吡咯的预期结构。
实施例3-采用在作为碳同素异形体的炭黑上负载的反应物合成1-己基-2,5-二甲基-1h-吡咯
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
向配备有磁搅拌器的100-ml单颈烧瓶中引入3.75g(0.151mol)己胺,4.25g(0.151mol)的2,5-己二酮和10g炭黑。该烧瓶配备有磁搅拌器并使混合物在150℃的温度下进行缓慢旋转2小时。然后使该反应混合物达到室温。取出固体样品并置于试管中。在室温下添加氘代氯仿(cdcl3)。室温下手动搅拌试管2分钟。倾析试管内包含的悬浮液。通过1h-nmr光谱分析这一液体,其揭示与图2中所示相同的峰,并因此证实了化合物1-己基-2,5-二甲基-1h-吡咯的预期结构。
实施例4-炭黑与1-己基-2,5-二甲基-1h-吡咯的加合物
所使用的己基吡咯如实施例1中合成。
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
向250-ml单颈烧瓶中引入10g炭黑和100ml丙酮。在超声浴中超声处理该悬浮液15分钟。在这一时间之后,添加2.33g己基吡咯在20ml丙酮中的溶液。超声处理所得悬浮液另外15分钟。在减压下去除溶剂。获得由炭黑和所吸收的己基吡咯组成的粉末。
将0.300g粉末置于配备有磁搅拌器的30-ml小瓶中。在180℃的温度下加热该反应混合物2小时。
在这一时间之后,冷却粉末到25℃。将该粉末置于具有隔膜的büchner中,并用蒸馏水反复洗涤。滤液是无色的。通过uv光谱分析洗涤水。
在所指定的热处理时间和所阐述的洗涤之后收集的加合物样品通过ft-ir分析表征,该分析通过在kbr中制备加合物样品的压片进行。图3中的ir光谱示出了在2900cm-1和2830cm-1处基团(ch2)序列典型的峰-在起始同素异形体的ir光谱中不存在的峰。
实施例5-石墨与1-己基-2,5-二甲基-1h-吡咯的加合物
所使用的己基吡咯如实施例1中合成。
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向250-ml单颈烧瓶中引入10g石墨和100ml丙酮。在超声浴中超声处理该悬浮液15分钟。在这一时间之后,添加2.33g己基吡咯在20ml丙酮中的溶液。超声处理所得悬浮液另外15分钟。在减压下去除溶剂。获得由石墨和所吸收的己基吡咯组成的粉末。
将0.300g粉末置于配备有磁搅拌器的30-ml小瓶中。在180℃的温度下加热该反应混合物2小时。
在这一时间之后,冷却粉末到25℃。将该粉末置于具有隔膜的büchner中,并用蒸馏水反复洗涤。滤液是无色的。通过uv光谱分析洗涤水。
通过在kbr中制备加合物样品的压片进行的ft-ir分析,表征在所指定的热处理时间和洗涤之后收集的加合物样品。
实施例6-合成1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷(六亚甲基双吡咯,hbp)
经验式:c18h28n2
摩尔质量:272.43
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(8.6mmol)六亚甲基二胺和1.96g(17.2mmol)的2,5-己二酮。在150℃下搅拌该混合物4小时。
以暗黄色油状物形式分离该产物,产率为80%并通过与质谱相连的气相色谱(gc-ms)和核磁共振(nmr)分析。图4示出了与质谱相连的色谱图。通过这一实验测定的质量对应于化合物1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷的理论质量。
实施例7-采用在作为碳同素异形体的石墨上负载的反应物合成1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(8.6mmol)六亚甲基二胺,1.96g(17.2mmol)的2,5-己二酮和3g石墨。该烧瓶配备有磁搅拌器并使混合物在150℃的温度下进行缓慢旋转2小时。然后使该反应混合物达到室温。取出固体样品并置于试管中。在室温下添加氘代氯仿(cdcl3)。室温下手动搅拌试管2分钟。倾析试管内包含的悬浮液。通过1h-nmr光谱分析这一液体。
实施例8-采用在作为碳同素异形体的炭黑上负载的反应物合成1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(8.6mmol)六亚甲基二胺,1.96g(17.2mmol)的2,5-己二酮和3g炭黑。该烧瓶配备有磁搅拌器并使混合物在150℃的温度下进行缓慢旋转2小时。然后使该反应混合物达到室温。取出固体样品并置于试管中。在室温下添加氘代氯仿(cdcl3)。室温下手动搅拌试管2分钟。倾析试管内包含的悬浮液。通过1h-nmr光谱分析这一液体。
实施例9-纳米石墨与1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷的加合物
所使用的吡咯化合物(六亚甲基双吡咯)如实施例6中合成。
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向250-ml单颈烧瓶中引入10g石墨和100ml丙酮。在超声浴中超声处理该悬浮液15分钟。在这一时间之后,添加2g六亚甲基双吡咯在20ml丙酮中的溶液。超声处理所得悬浮液另外15分钟。在减压下去除溶剂。获得由石墨和所吸收的六亚甲基双吡咯组成的粉末。
将0.300g粉末置于配备有磁搅拌器的30-ml小瓶中。在180℃的温度下加热该反应混合物2小时。
在这一时间之后,冷却粉末到25℃。将该粉末置于具有隔膜的büchner中,并用蒸馏水反复洗涤。滤液是无色的。通过uv光谱分析洗涤水。
通过在kbr中制备加合物样品的压片进行的ft-ir分析,表征在所指定的热处理时间和所阐述的洗涤之后收集的加合物样品。图5中的ir光谱示出了在2910cm-1和2840cm-1处基团(ch2)序列典型的峰-在起始同素异形体的ir光谱中不存在的峰。
实施例10-炭黑与1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷的加合物
所使用的吡咯化合物(六亚甲基双吡咯)如实施例6中合成。
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
向250-ml单颈烧瓶中引入10g炭黑和100ml丙酮。在超声浴中超声处理该悬浮液15分钟。在这一时间之后,添加2g六亚甲基双吡咯在20ml丙酮中的溶液。超声处理所得悬浮液另外15分钟。在减压下去除溶剂。获得由炭黑和所吸收的六亚甲基双吡咯组成的粉末。
将0.300g粉末置于配备有磁搅拌器的30-ml小瓶中。在180℃的温度下加热该反应混合物2小时。
在这一时间之后,冷却粉末到25℃。将该粉末置于具有隔膜的büchner中,并用蒸馏水反复洗涤。滤液是无色的。通过uv光谱分析洗涤水。
通过在kbr中制备加合物样品的压片进行的ft-ir分析,表征在所指定的热处理时间和所阐述的洗涤之后收集的加合物样品。
实施例11-合成2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯(吡咯基丙基三甲氧基硅烷(pptms)
经验式:c12h23no3si
摩尔质量:257.14
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(5.58mmol)的3-(三甲氧基甲硅烷基)丙-1-胺和0.640g(5.58mmol)的2,5-己二酮。在150℃下搅拌该混合物6小时。以强烈的浅黄色粘性固体形式分离产物并通过与质谱相连的气相色谱(gc-ms)和核磁共振(nmr)分析。gc-ms分析示出了化合物2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯以及未反应的3-(三甲氧基甲硅烷基)丙-1-胺。然后将该黄色固体溶解在二氯甲烷中。用去离子水洗涤所得溶液。在na2so4上干燥有机相,并通过减压彻底干燥。所分离的固体是纯化合物2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯。这一化合物的重量允许我们计算产率等于89%。
图6示出了1h-nmr光谱。
实施例12-采用在作为碳同素异形体的石墨上负载的反应物合成2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(5.58mmol)的3-(三甲氧基甲硅烷基)丙-1-胺,0.640g(5.58mmol)的2,5-己二酮和3g石墨。该烧瓶配备有磁搅拌器并使混合物在150℃的温度下进行缓慢旋转2小时。然后使该反应混合物达到室温。取出固体样品并置于试管中。在室温下添加氘代氯仿(cdcl3)。室温下手动搅拌试管2分钟。倾析试管内包含的悬浮液。通过1h-nmr光谱分析这一液体,其揭示了与图6中所示相同的峰,并因此证实化合物2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯的预期结构。
实施例13-采用在作为碳同素异形体的炭黑上负载的反应物合成2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(5.58mmol)的3-(三甲氧基甲硅烷基)丙-1-胺,0.640g(5.58mmol)of2,5-己二酮和3gof炭黑。该烧瓶配备有磁搅拌器并使混合物在150℃的温度下进行缓慢旋转2小时。然后使该反应混合物达到室温。取出固体样品并置于试管中。在室温下添加氘代氯仿(cdcl3)。室温下手动搅拌试管2分钟。倾析试管内包含的悬浮液。通过1h-nmr光谱分析这一液体,其揭示了与图6中所示相同的峰,并因此证实化合物2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯的预期结构。
实施例14-石墨与2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯的加合物
所使用的吡咯化合物如实施例11中合成。
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向250-ml单颈烧瓶中引入10g石墨和100ml丙酮。在超声浴中超声处理该悬浮液15分钟。在这一时间之后,添加2g六亚甲基双吡咯在20ml丙酮中的溶液。超声处理所得悬浮液另外15分钟。在减压下去除溶剂。获得由石墨和所吸收的2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯组成的粉末。
将0.300g粉末置于配备有磁搅拌器的30-ml小瓶中。在180℃的温度下加热该反应混合物2小时。
在这一时间之后,冷却粉末到25℃。将该粉末置于具有隔膜的büchner中,并用去离子水洗涤3次。滤液是无色的。通过uv光谱分析来自第四次的水:没有检测到吸收。
通过在kbr中制备加合物样品的压片进行的ft-ir分析,表征在所指定的热处理时间和所阐述的洗涤之后收集的加合物样品。图7中的ir光谱示出了在2910cm-1和2827cm-1处基团(ch2)序列典型的峰-在起始同素异形体的ir光谱中不存在的峰。
实施例15-炭黑与2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯的加合物
所使用的吡咯化合物如实施例11中合成。
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
向250-ml单颈烧瓶中引入10g炭黑和100ml丙酮。在超声浴中超声处理该悬浮液15分钟。在这一时间之后,添加2g六亚甲基双吡咯在20ml丙酮中的溶液。超声处理所得悬浮液另外15分钟。在减压下去除溶剂。获得由炭黑和所吸收的2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯组成的粉末。
将0.300g粉末置于配备有磁搅拌器的30-ml小瓶中。在180℃的温度下加热该反应混合物2小时。
在这一时间之后,冷却粉末到25℃。将该粉末置于具有隔膜的büchner中,并用去离子水洗涤3次。滤液是无色的。通过uv光谱分析来自第四次洗涤的水:没有检测到吸收。
实施例16-合成3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺(二甲基胺丙烷吡咯dapp)
经验式:c11h20n2
摩尔质量:180.16
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(9.7mmol)的n1,n1-二甲基丙烷-1,3-二胺和1.10g(9.7mmol)的2,5-己二酮。在150℃下搅拌该混合物6小时。以琥珀色油状物形式分离该产物并通过核磁共振(nmr)分析,其显示出仅仅预期的化合物3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺。通过nmr分析所发现的重量和观察到的化学纯度允许评价产率等于95%。图8示出了1h-nmr光谱。
实施例17-采用在作为碳同素异形体的石墨上负载的反应物合成3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(9.7mmol)的n1,n1-二甲基丙烷-1,3-二胺,1.10g(9.7mmol)的2,5-己二酮和3g石墨。该烧瓶配备有磁搅拌器并使混合物在150℃的温度下进行缓慢旋转2小时。然后使该反应混合物达到室温。取出固体样品并置于试管中。在室温下添加乙酸乙酯。室温下手动搅拌试管2分钟。倾析试管内包含的悬浮液。通过采用乙酸乙酯/己烷溶剂混合物(1/9)进行的薄层色谱法,分析上清液。uv分析显示出具有与实施例16中获得的物质3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺和用乙酸乙酯从石墨中提取的物质相同rf(0.6)的渗开圆点,正如刚才所描述的。
实施例18-采用在作为碳同素异形体的炭黑上负载的反应物合成3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(9.7mmol)的n1,n1-二甲基丙烷-1,3-二胺,1.10g(9.7mmol)的2,5-己二酮和3g炭黑。该烧瓶配备有磁搅拌器并使混合物在150℃的温度下进行缓慢旋转2小时。然后使该反应混合物达到室温。取出固体样品并置于试管中。在室温下添加乙酸乙酯。室温下手动搅拌试管2分钟。倾析试管内包含的悬浮液。通过采用乙酸乙酯/己烷溶剂混合物(1/9)进行的薄层色谱法,分析上清液。uv分析显示出具有与实施例16中获得的物质3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺和用乙酸乙酯从石墨中提取的物质相同rf(0.6)的渗开圆点,正如刚才所描述的。
实施例19-石墨与3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺(dapp)的加合物
所使用的吡咯化合物如实施例16中合成。
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向250-ml单颈烧瓶中引入10g石墨和100ml丙酮。在超声浴中超声处理该悬浮液15分钟。在这一时间之后,添加2g的dapp在20ml丙酮中的溶液。超声处理所得悬浮液另外15分钟。在减压下去除溶剂。获得由石墨和所吸收的dapp组成的粉末。
将0.300g粉末置于配备有磁搅拌器的30-ml小瓶中。在180℃的温度下加热该反应混合物2小时。
在这一时间之后,冷却粉末到25℃。将该粉末置于具有隔膜的büchner中,并用去离子水洗涤3次。滤液是无色的。通过uv光谱分析来自第四次洗涤的水:没有检测到吸收。
实施例20-合成o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇(吡咯基peg,ppeg)
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(1.6mmol)的o-(2-氨基丙基)-o'-(2-甲氧基乙基)聚丙二醇(mn=600)和0.190g(1.6mmol)的2,5-己二酮。在150℃下搅拌该混合物6小时。以非常粘稠的琥珀色液体形式分离该产物并通过核磁共振(nmr)分析,其显示出仅仅预期的化合物o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇。通过nmr分析所发现的重量和观察到的化学纯度允许评价产率等于97%。图9示出了1h-nmr光谱。
实施例21-采用在作为碳同素异形体的石墨上负载的反应物合成o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向配备有磁搅拌器的100-ml单颈烧瓶中引入1g(1.6mmol)的o-(2-氨基丙基)-o'-(2-甲氧基乙基)聚丙二醇,0.190g(1.6mmol)的2,5-己二酮和3g石墨。
实施例22-作为碳同素异形体的石墨与o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇的加合物
所使用的吡咯化合物如实施例19中合成。
所使用的石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
向250-ml单颈烧瓶中引入10g石墨和100ml丙酮。在超声浴中超声处理该悬浮液15分钟。在这一时间之后,添加2g的o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇在20ml丙酮中的溶液。超声处理所得悬浮液另外15分钟。在减压下去除溶剂。获得由石墨和所吸收的o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇组成的粉末。
将0.300g粉末置于配备有磁搅拌器的30-ml小瓶中。在180℃的温度下加热该反应混合物2小时。
在这一时间之后,冷却粉末到25℃。
实施例23-26-吡咯化合物与石墨的加合物在乙酸乙酯中分散体的稳定性试验
石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
这一试验的目的是验证由碳同素异形体、石墨和吡咯化合物组成的加合物形成随着时间流逝稳定的悬浮液的能力。在有机分子,例如乙酸乙酯中评价在溶剂中的稳定性。
工序:
将10mg粉化加合物置于10-ml烧瓶中并添加乙酸乙酯(10ml)。在2升超声浴中,采用260w的功率,超声处理该混合物20分钟。
使用pasteur移液管,将浓度为1mg/ml的加合物悬浮液(3ml)转移到具有1cm光路长度的石英比色皿(体积1或3ml)中并使用uv-vis分光光度计分析。该仪器事先用纯溶剂重置,记录uv光谱(200-340nm)。该uv-可见光谱示出吸收强度作为200至750nm的辐射波长的函数。
为了评价所得悬浮液随着时间流逝的稳定性,然后1周后反复uv-vis吸收测量。
表1中给出了稳定性试验结果。
表1.吡咯化合物与石墨的加合物在乙酸乙酯中分散体的稳定性试验a
a石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
bhp=1-己基-2,5-二甲基-1h-吡咯(己基吡咯)
chbp=1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷(六亚甲基双吡咯)
dpptms=2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯(吡咯基丙基三甲氧基硅烷)
eppeg=o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇(吡咯基peg)
实施例27-29-吡咯化合物与炭黑的加合物在乙酸乙酯中分散体的稳定性试验
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
这一试验的目的是验证由碳同素异形体、炭黑和吡咯化合物组成的加合物形成随着时间流逝稳定的悬浮液的能力。在有机分子,例如乙酸乙酯中评价在溶剂中的稳定性。
工序:
将10mg粉化加合物置于10-ml烧瓶中并添加乙酸乙酯(10ml)。在2升超声浴中,采用260w的功率,超声处理该混合物20分钟。
使用pasteur移液管,将浓度为1mg/ml的加合物悬浮液(3ml)转移到具有1cm光路长度的石英比色皿(体积1或3ml)中并使用uv-vis分光光度计分析。该仪器事先用纯溶剂重置,记录uv光谱(200-340nm)。该uv-可见光谱示出吸收强度作为200至750nm的辐射波长的函数。
为了评价所得悬浮液随着时间流逝的稳定性,然后1周后反复uv-vis吸收测量。
表2中给出了稳定性试验结果。
表2.吡咯化合物与炭黑的加合物在乙酸乙酯中分散体的稳定性试验a
a所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
bhp=1-己基-2,5-二甲基-1h-吡咯(己基吡咯)
chbp=1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷(六亚甲基双吡咯)
dpptms=2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯(吡咯基丙基三甲氧基硅烷)
实施例30-34-吡咯化合物与石墨的加合物在正己烷中分散体的稳定性试验
石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
这一试验的目的是验证由碳同素异形体、石墨和吡咯化合物组成的加合物形成随着时间流逝稳定的悬浮液的能力。在非极性介质,正己烷中评价在溶剂中的稳定性。
工序:
将10mg粉化加合物置于10-ml烧瓶中并添加正己烷(10ml)。在2升超声浴中,采用260w的功率,超声处理该混合物20分钟。
使用pasteur移液管,将浓度为1mg/ml的加合物悬浮液(3ml)转移到具有1cm光路长度的石英比色皿(体积1或3ml)中并使用uv-vis分光光度计分析。该仪器事先用纯溶剂重置,记录uv光谱(200-340nm)。该uv-可见光谱示出吸收强度作为200至750nm的辐射波长的函数。
为了评价所得悬浮液随着时间流逝的稳定性,然后1周后反复uv-vis吸收测量。
表3中给出了稳定性试验结果。
表3.吡咯化合物与石墨的加合物在正己烷中分散体的稳定性试验a
a石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
bhp=1-己基-2,5-二甲基-1h-吡咯(己基吡咯)
chbp=1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷(六亚甲基双吡咯)
dpptms=2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯(吡咯基丙基三甲氧基硅烷)
edapp=3-(2,5-二甲基-1h-吡咯-1-基)-n,n-二甲基丙-1-胺(二甲基胺丙烷吡咯)
eppeg=o-(2-(2,5-二甲基吡咯-1-基)丙基)-o'-(2-甲氧基乙基)聚丙二醇(吡咯基peg)
实施例35-37-吡咯化合物与炭黑的加合物在正己烷中分散体的稳定性试验
所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
这一试验的目的是验证由碳同素异形体、炭黑和吡咯化合物组成的加合物形成随着时间流逝稳定的悬浮液的能力。在非极性介质,正己烷中评价在溶剂中的稳定性。
工序:
将10mg粉化加合物置于10-ml烧瓶中并添加正己烷(10ml)。在2升超声浴中,采用260w的功率,超声处理该混合物20分钟。
使用pasteur移液管,将浓度为1mg/ml的加合物悬浮液(3ml)转移到具有1cm光路长度的石英比色皿(体积1或3ml)中并使用uv-vis分光光度计分析。该仪器事先用纯溶剂重置,记录uv光谱(200-340nm)。该uv-可见光谱示出吸收强度作为200至750nm的辐射波长的函数。
为了评价所得悬浮液随着时间流逝的稳定性,然后1周后反复uv-vis吸收测量。
表4中给出了稳定性试验结果。
表4.吡咯化合物与炭黑的加合物在正己烷中分散体的稳定性试验a
a所使用的炭黑是具有下述特性的炭黑n326(cb)(cabot):球形颗粒的平均直径为30nm,表面积等于77m2/g(通过氮气吸附测定),dbp吸收等于85ml/100g。
bhp=1-己基-2,5-二甲基-1h-吡咯(己基吡咯)
chbp=1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷(六亚甲基双吡咯)
dpptms=2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯(吡咯基丙基三甲氧基硅烷)
实施例38-40-吡咯化合物与石墨的加合物在甲苯中分散体的稳定性试验
石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
这一试验的目的是验证由碳同素异形体、石墨和吡咯化合物组成的加合物形成随着时间流逝稳定的悬浮液的能力。在有机溶剂,例如甲苯中评价在溶剂中的稳定性。
工序:
将10mg粉化加合物置于10-ml烧瓶中并添加甲苯(10ml)。在2升超声浴中,采用260w的功率,超声处理该混合物20分钟。
使用pasteur移液管,将浓度为1mg/ml的加合物悬浮液(3ml)转移到具有1cm光路长度的石英比色皿(体积1或3ml)中并使用uv-vis分光光度计分析。该仪器事先用纯溶剂重置,记录uv光谱(200-340nm)。该uv-可见光谱示出吸收强度作为200至750nm的辐射波长的函数。
为了评价所得悬浮液随着时间流逝的稳定性,然后1周后反复uv-vis吸收测量。
表5中给出了稳定性试验结果。
表5.吡咯化合物与石墨的加合物在甲苯中分散体的稳定性试验a
a石墨是获自asburygraphitemillsinc.的合成石墨8427,其具有99.8wt.%的最小碳含量和330m2/g的表面积。
bhp=1-己基-2,5-二甲基-1h-吡咯(己基吡咯)
chbp=1,6-双(2,5-二甲基-1h-吡咯-1-基)己烷(六亚甲基双吡咯)
dpptms=2,5-二甲基-1-(3-(三甲氧基甲硅烷基)丙基)-1h-吡咯(吡咯基丙基三甲氧基硅烷)。
本文地址:https://www.jishuxx.com/zhuanli/20240726/121173.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。