基因序列压缩方法、检索方法及装置
- 国知局
- 2024-11-06 14:22:52
本发明属于生物信息学领域,具体涉及基因序列压缩方法、检索方法及装置。
背景技术:
1、基因序列检索是生物信息学中的关键技术,用于在庞大的基因组数据库中查找特定基因或dna序列。通过比对算法,研究人员可以找到序列之间的相似性,从而揭示其功能和进化关系。常用的序列检索工具包括blast、fasta和smith-waterman算法。其中,blast因其速度快、灵敏度高而被广泛应用,而smith-waterman算法则以高精度著称,适用于小规模比对。此外,隐马尔可夫模型(hmm)在蛋白质序列比对和结构预测中也发挥重要作用。
2、基因序列检索在功能注释、进化研究、疾病研究和药物开发等多个领域中具有重要应用。通过比对新测序的基因或蛋白质序列与已知数据库中的序列,研究人员可以推测其功能,帮助了解基因的生物学作用和分子机制。比较不同物种间的基因序列有助于研究其进化关系,而在疾病研究中,基因序列检索可以帮助发现和诊断与疾病相关的突变基因。在药物开发中,基因序列检索有助于识别疾病相关的基因或蛋白质,指导新药的开发。
3、随着测序技术和计算能力的提升,基因序列检索方法不断进步。大数据和人工智能技术的引入为基因序列的快速、精准比对提供了新的解决方案。未来,基因序列检索将更加高效、智能,在科学研究和实际应用中发挥更大的作用。通过不断优化算法和引入新技术,基因序列检索不仅推动了基因组学研究的进步,也在医学和农业等领域产生深远影响。
技术实现思路
1、本发明的目的是提供基因序列压缩方法、检索方法及装置,提高对基因数据的处理规模和检索效率。
2、本发明实现上述目的所采用的技术方案如下:
3、第一方面,本发明提供一种基因序列压缩方法,包括以下步骤:
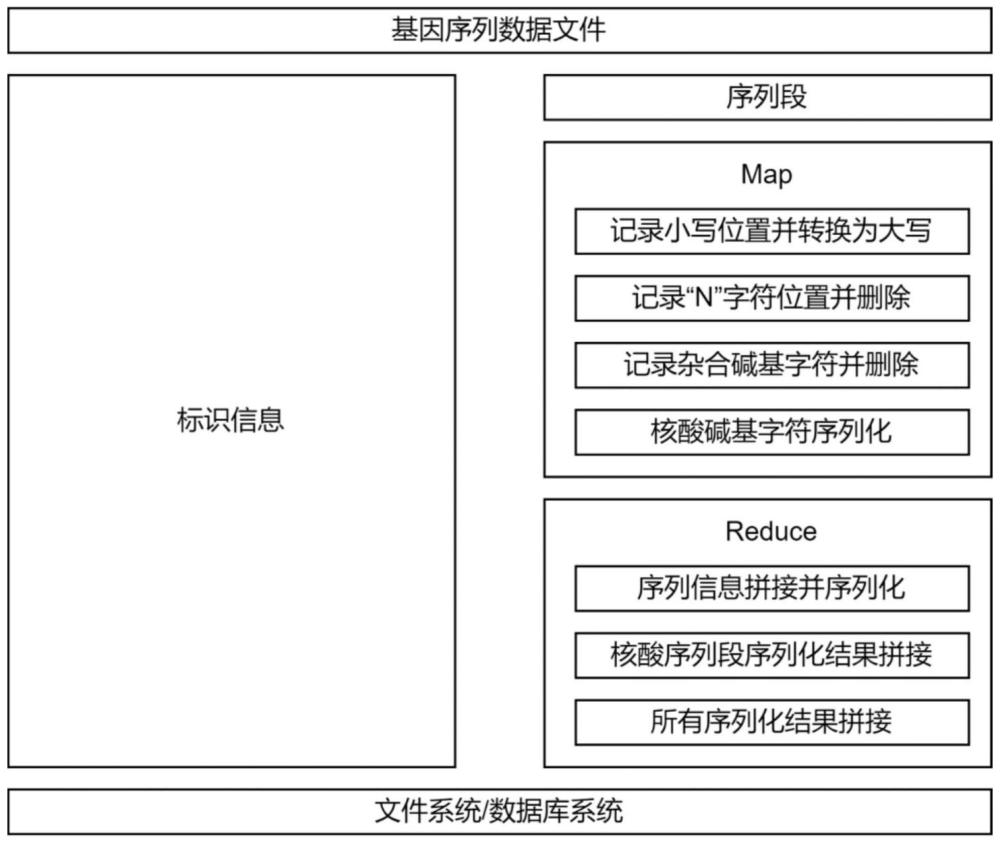
4、对基因序列文件解析得到序列信息和序列段;
5、基于map操作将序列段处理成仅有核酸碱基的序列并进行序列化;
6、基于reduce操作将上述序列信息和序列化结果进行拼接并存储到序列文件中。
7、进一步地,读取基因序列文件中的标识行,得到序列信息。
8、进一步地,针对基因序列文件中的原始序列,去除空白字符并以64kb为一段进行划分,其中末尾不足64kb的部分划分到最后一个段,得到序列段。
9、进一步地,对于包含多条序列数据的基因序列文件,根据标识行将基因序列文件拆分成仅包含单条序列数据的基因序列文件,然后针对拆分后的每个基因序列文件进行解析,得到对应的序列信息和序列段。
10、进一步地,基于map操作将序列段处理成仅有核酸碱基的序列并进行序列化的步骤包括:
11、记录序列段中的小写字符的相对位置和长度,并以元组形式存放到列表中,再将小写字符转换为大写字符;
12、记录序列段中的未知碱基字符的相对位置和长度,并以元组形式存放到列表中,再删除所有未知碱基字符;
13、记录序列段中的杂合碱基的字符和相对位置,并以元组形式存放到列表中,再删除所有的杂合碱基;
14、通过上述步骤处理后得到仅有核酸碱基的序列,并对仅有核酸碱基的序列中的每个核酸碱基按照a-00,g-01,c-10,t-11或u-11的关系进行映射,并将每4个碱基的映射结果打包为一个字节,实现对仅有核酸碱基的序列的序列化。
15、进一步地,基于reduce操作将序列信息和序列化结果进行拼接的步骤包括:
16、1)将每个序列段的序列信息进行拼接,再进行序列化;
17、2)将核酸碱基序列的序列化结果进行拼接;
18、3)将步骤1)得到的序列化结果和步骤2)得到的拼接结果进行拼接。
19、第二方面,本发明提供一种基因序列检索方法,包括以下步骤:
20、根据用户提供的目标序列信息,从存储的序列文件中读取目标序列的数据流;
21、对用户提供的查询序列进行预处理;
22、根据预处理结果计算距离表;
23、根据距离表在目标序列中对查询序列进行匹配,并返回匹配结果。
24、进一步地,对用户提供的查询序列进行预处理的步骤包括
25、基于map操作将查询序列进行序列化;
26、在序列化结果前依次添加1至3个腺嘌呤字符,得到3个移位序列化结果;
27、将原始的查询序列和移位序列化结果转换为字节数组。
28、进一步地,根据预处理结果计算距离表的方法为将原始查询序列和移位序列化结果转换的字节数组进行处理得到距离表,该距离表中的每行包括一个字节值和一个相对距离列表,处理步骤包括:
29、创建一个位置信息列表,并将所有元素初始化,同时初始化遍历索引index;
30、对于字节数组白每个index对应的值,同时计算当前字节在位置信息列表中的对应索引;每个字节添加偏移值,使字节范围映射到位置信息列表的长度范围上;
31、检查位置信息列表中的对应索引是否为初始值,如果是,则在相对距离列表中添加相应行;否则计算当前索引与位置信息列表中的对应索引之间的距离,并找到当前字节对应的相对距离列表,将距离插入到相对距离列表的末尾;
32、更新当前字节最近出现的位置,将位置信息列表中的对应索引更新为当前索引index。
33、进一步地,根据距离表在目标序列中对查询序列进行匹配的步骤包括:
34、初始化一个长度与查询序列的字节数组相同的结果列表和匹配计数器;
35、遍历目标序列字节数组中的值,当其值在距离表中时,取出距离表中的相对距离列表,依次比对相对位置上的值是否相同,相同则存入结果列表,并令结果计数加一;
36、当结果计数等于或超过与查询字节数组长度的一半时,即可判断匹配成功,返回当前结果列表第一个元素在查询序列字节数组的位置;否则初始化结果列表和结果计数,并使遍历索引向前移动至当前索引加上查询字节数组长度,然后重复遍历操作。
37、一种基因序列的存储和检索装置,包括:
38、基因序列导入模块,用于从用户指定路径导入基因序列文件到数据库或文件系统中;
39、基因序列导出模块,用于从数据库或文件系统导出用户指定的基因序列到指定路径;
40、基因序列查询模块,用于接收用户输入的目标序列和查询序列信息,返回在数据库或文件系统中的查询结果;
41、功能拓展模块,用于实现用户针对基因序列信息相关的自定义函数功能。
42、进一步地,自定义函数包括以下函数中的一种或多种:
43、biosequence.fromurl:从统一资源定位符中导入基因序列;
44、biosequence.frombytes:从字节码中导入基因序列;
45、biosequence.fromfile:从指定文件路径中导入基因序列;
46、biosequence.exportsequence:从图数据库中导出基因序列到指定路径;
47、biosequence.getinformation:获取基因序列文件的信息,包括提交机构、提交时间信息;
48、biosequence.length:获取基因序列的长度;
49、biosequence.getlowercaseposition:获取小写字符相对位置和长度;
50、biosequence.getncaseposition:获取未知碱基字符的起始点和长度;
51、biosequence.getnonagctcaseposition:获取杂合碱基的起始点和字符;
52、biosequence.searchone:检索目标序列中首次出现的查询序列,返回两条序列的比对结果;
53、biosequence.searchall:检索目标序列中所有出现的查询序列,返回所有相似序列的比对结果。
54、本发明的创新点在于:
55、1.实现了基于mapreduce的2bit基因序列压缩算法:在分布式系统中实现了一种新的基因序列压缩方法,采用mapreduce框架,使得算法能够处理大规模基因数据。
56、2.新的基因序列检索方式:提出了一种新的检索方法,与传统方法不同,在压缩数据中直接进行匹配,提高了检索效率。
57、3.数据库系统中的应用装置:实现了上述检索方式在数据库系统中的应用,使得基因数据的存储和检索更加高效。
58、本发明的优点在于:
59、1.优化的2bit压缩算法:a)压缩速度提升:使用mapreduce方法,对2bit算法进行了优化,使得压缩速度高于传统实现。b)保留基因序列特征:使用2bit压缩方法,保留了部分基因序列特征,为后续检索提供了基础。
60、2.新的检索算法:a)字节流匹配:在字节流中进行匹配,不需要将压缩文件还原成字符串,从而避免了传统方法中需要完全还原压缩文件再进行匹配的问题。b)无索引检索:不需要构建字符串索引,直接在字节流中进行检索,减少了基因序列检索的时间。c)高效的匹配结果处理:在字符串还原时,使用匹配结果中的字节作为种子,仅需解压部分目标序列数据,降低了传统方法中基于种子扩展算法的计算时间。
61、基于上述创新点和优点,本发明在基因序列的压缩和检索方面提供了更高效的解决方案。
本文地址:https://www.jishuxx.com/zhuanli/20241106/321699.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表