一种电解法制备2
‑
乙酰基吡嗪的方法
技术领域
1.本发明属于精细化工和有机电化学合成技术领域,具体的说,涉及一种电解法制备2
‑
乙酰基吡嗪的方法。
背景技术:
2.乙酰基吡嗪是一种应用广泛的食品添加剂和医药中间体。目前,制备方法主要为有机金属试剂法,将吡嗪酰胺脱水成相应的腈,同格氏试剂甲基溴化镁反应,再水解得到乙酰基吡嗪,其最大的缺点是反应须在低温下进行且成本高。为了降低成本,人们尝试过渡金属盐活化过硫酸铵的方法,过渡金属盐分为贵金属盐和非贵金属盐两类。贵金属盐一般采用银盐,它们在合成2
‑
乙酰基吡嗪时具有良好的效果,但仍没有改变成本高、难回收的缺点。而对于非贵金属盐,一般使用亚铁盐,其价格便宜、收率高,但实际生产中,副产物多,难分离,降低了产品纯度。同时,如果产品中亚铁盐没有除尽,产品性状易改变,产品质量低。
3.有机电化学技术具有工艺流程简单,反应条件温和,反应易控制、环境友好等优点。
4.有鉴于此,有必要发展一种以电化学技术为主制备2
‑
乙酰基吡嗪的方法。
技术实现要素:
5.本发明的目的是提供一种电解法制备2
‑
乙酰基吡嗪的方法。
6.为了实现上述目的,本发明采用的技术方案如下:
7.本发明的第一方面提供了一种电解法制备2
‑
乙酰基吡嗪的方法,包括以下步骤:
8.采用h型电解槽作为反应器,吡嗪和丙酮酸溶于二氯甲烷中,氧化剂过硫酸铵溶于水中,加入阴极室中,再添加二甲基亚砜;
9.所述吡嗪和丙酮酸的摩尔比为(1~2):1,所述丙酮酸溶于二氯甲烷的浓度为0.2~1mol
·
l
‑1,氧化剂过硫酸铵溶于水的浓度为1~1.5mol
·
l
‑1,所述二甲基亚砜的质量为阴极室中阴极液总质量的1%~10%;
10.阳极室中加入质量分数为5~30%(优选为20%)的硫酸溶液;保持恒温(温度优选为25℃),通电电解,保持电流密度为10~800a
·
m
‑2,通电量达到2f
·
mol
‑1后停止通电,分出有机相,得到含有2
‑
乙酰基吡嗪的二氯甲烷溶液,2
‑
乙酰基吡嗪收率以吡嗪计。
11.所述h型电解槽包括左右设置的阳极室和阴极室,所述阳极室内设有阳极板,所述阴极室内设有阴极板,所述阳极板和所述阴极板的上部通过直流电源连通,所述阳极室和所述阴极室的中间部分通过阳离子交换膜连通,所述阴极室内部设有端部延伸至外部的温度计,所述阳极室的内底部设有第二搅拌子,在所述阳极室的下面设有第二磁力搅拌器,所述阴极室的内底部设有第一搅拌子,在所述阴极室的下面设有第一磁力搅拌器。
12.所述阳离子交换膜为阳离子交换膜nafion pfsa membranes(n
‑
324)。
13.所述阳极板为阳极镀钌钛板、石墨、镀铱钛网。
14.所述阴极板为阴极铅板。
15.优选的,所述丙酮酸溶于二氯甲烷的浓度为0.3~0.4mol
·
l
‑1,最优选为0.33mol
·
l
‑1。
16.优选的,所述氧化剂过硫酸铵溶于水的浓度为1.5mol
·
l
‑1。
17.优选的,所述二甲基亚砜的质量为阴极室中阴极液总质量的2~5%。
18.优选的,所述电流密度为100~800a
·
m
‑2。
19.由于采用上述技术方案,本发明具有以下优点和有益效果:
20.本发明提供的电解法制备2
‑
乙酰基吡嗪的方法反应条件温和,该方法在常温下即可反应;反应过程可控,通过控制电流密度的大小控制硫酸根自由基的生成速率,实现原料的最大化利用;该反应几乎没有副产物产生,2
‑
乙酰基吡嗪易分离,产品纯度高;剩余的水相反应液可回收重复利用。
21.本发明提供的电解法制备2
‑
乙酰基吡嗪的方法,以吡嗪和丙酮酸为原料电解制备2
‑
乙酰基吡嗪,该工艺在h型电解槽中进行,阴极室为含有原料和过硫酸铵的水/二氯甲烷两相溶液,阳极室为硫酸溶液。过硫酸铵在阴极还原生成的硫酸根自由基与原料反应,2
‑
乙酰基吡嗪的收率可达到41%。该工艺在常温常压下反应,以电子作为硫酸根自由基的引发剂,避免金属催化剂的使用,副产物少。反应后的水相为仅含有部分原料和硫酸铵的溶液,可通过萃取和氧化硫酸铵实现原料和过硫酸铵的回收利用,绿色环保、操作简便,适用于工业化生产。
22.格氏试剂法是合成2
‑
乙酰基吡嗪的常用方法,而本发明以价格低廉的过硫酸铵为氧化剂,在电解的条件下活化过硫酸铵,该过程电子充当活化剂,环境友好,仅一步就能合成乙酰基吡嗪。反应结束后,反应液中仅含有吡嗪和2
‑
乙酰基吡嗪,可通过简单的蒸馏分离。解决了现有技术中制备2
‑
乙酰基吡嗪工艺中存的生产成本高、产品质量低及污染环境的技术问题。
23.本发明在阴极室,过硫酸根还原为硫酸根自由基,硫酸根自由基具有更强的氧化性,氧化丙酮酸生成乙酰基自由基。由于过硫酸铵容易被直接还原为硫酸根且硫酸根自由基不能稳定存在,因此可通过控制电流密度,控制硫酸根自由基的生成速率,从而控制乙酰基自由基的生成速率,防止副产物的生成,也能防止大量硫酸根自由基降解吡嗪和乙酰基吡嗪,具有操控性好,控制精准的特点。由于加入的吡嗪和过硫酸铵是过量的,反应结束后,水相中仍含有大量吡嗪和过硫酸铵,可加入适量原料继续反应重复利用。
附图说明
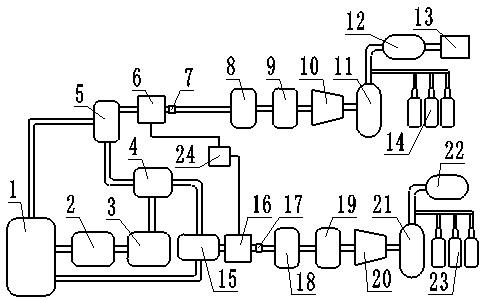
24.图1是h型电解槽电解装置的示意图。
25.1为直流电源、2为阳极板、3为阳极室、4为温度计、5为阴极板、61为第一搅拌子、62为第二搅拌子、71为第一磁力搅拌器、72为第二磁力搅拌器、8为阳离子交换膜、9为阴极室。
具体实施方式
26.为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
27.所述h型电解槽包括左右设置的阳极室3和阴极室9,所述阳极室3内设有阳极板2,
所述阴极室9内设有阴极板5,所述阳极板2和所述阴极板5的上部通过直流电源1连通,所述阳极室3和所述阴极室9的中间部分通过阳离子交换膜8连通,所述阴极室9内部设有端部延伸至外部的温度计4,所述阳极室3的内底部设有第二搅拌子62,在所述阳极室3的下面设有第二磁力搅拌器72,所述阴极室9的内底部设有第一搅拌子61,在所述阴极室9的下面设有第一磁力搅拌器71。
28.所述阳离子交换膜8为阳离子交换膜nafion pfsa membranes(n
‑
324)。
29.所述阳极板2为阳极镀钌钛板、石墨、镀铱钛网。
30.所述阴极板5为阴极铅板。
31.本发明2
‑
乙酰基吡嗪的制备方法有关电化学反应方程式如下:
32.阳极:
33.2h2o
→
4h
o2 4e
‑
34.阴极:
[0035][0036]
生成的硫酸根自由基参与如下反应:
[0037][0038]
该装置的具体使用步骤如下:
[0039]
首先将溶剂二氯甲烷、吡嗪、丙酮酸、水、过硫酸铵加入阴极室,再添加二甲基亚砜,打开直流电源1,然后开通第一磁力搅拌器71,第一搅拌子61开始转动使得原料混合,在阳极室加入硫酸溶液,然后开通第二磁力搅拌器72,第二搅拌子62开始转动,阳离子交换膜8将阴阳两室隔开,仅有nh
4
和h
通过,形成完整的电流通路。阳极板2发生上述阳极反应,阴极板3发生上述阴极反应,控制电流密度和通电量,获得2
‑
乙酰基吡嗪。
[0040]
实施例1
[0041]
采用h型电解槽作为反应器,阳极镀钌钛板,阴极铅板,阳极室和阴极室用阳离子交换膜nafion pfsa membranes(n
‑
324)隔开。
[0042]
用二氯甲烷溶解0.067mol吡嗪、0.033mol丙酮酸定容至100ml,取100ml饱和过硫酸铵溶液,将上述溶液加入阴极室中,再添加5g二甲基亚砜。所述吡嗪和丙酮酸的物质的量比为2:1,所述丙酮酸溶于二氯甲烷的浓度为0.33mol
·
l
‑1,氧化剂过硫酸铵溶于水的浓度为1.5mol
·
l
‑1,所述二甲基亚砜的质量为阴极室中吡嗪/丙酮酸溶液和过硫酸铵溶液总质量的2%。
[0043]
阳极室中加入200ml质量分数20%的硫酸溶液;保持恒温25℃搅拌,通电电解,保持电流密度为100a
·
m
‑2,通电量达到2f
·
mol
‑1后停止通电,分出有机相,得到含有2
‑
乙酰基吡嗪的二氯甲烷溶液,2
‑
乙酰基吡嗪收率以吡嗪计;取样分析,采用上海天美公司gc
‑
7900型气相色谱仪,美国安捷伦公司tm
‑
5毛细管柱进行分析,色谱升温程序为:进样口温度250℃,检测器温度280℃,柱箱初始温度50℃,以10℃
·
min
‑1升温至250℃,保持5min;2
‑
乙酰基吡嗪的收率为40.2%,无副产物生成。
[0044]
实施例2
[0045]
采用h型电解槽作为反应器,阳极镀钌钛板,阴极铅板,阳极室和阴极室用阳离子交换膜nafion pfsa membranes(n
‑
324)隔开。
[0046]
用二氯甲烷溶解0.033mol吡嗪、0.033mol丙酮酸定容至100ml,取100ml饱和过流酸铵溶液,将上述溶液加入阴极室中,再添加5g二甲基亚砜。
[0047]
所述吡嗪和丙酮酸物质的量比为1:1,所述丙酮酸溶于二氯甲烷的浓度为0.33mol
·
l
‑1,氧化剂过硫酸铵溶于水的浓度为1.5mol
·
l
‑1,所述二甲基亚砜的质量为阴极室中吡嗪/丙酮酸溶液和过硫酸铵溶液总质量的2%;
[0048]
阳极室中加入200ml质量分数20%的硫酸溶液;保持恒温25℃搅拌,通电电解,保持电流密度为100a
·
m
‑2,通电量达到2f
·
mol
‑1后停止通电,分出有机相,得到含有2
‑
乙酰基吡嗪的二氯甲烷溶液,2
‑
乙酰基吡嗪收率以吡嗪计;取样分析,采用上海天美公司gc
‑
7900型气相色谱仪,美国安捷伦公司tm
‑
5毛细管柱进行分析,色谱升温程序为:进样口温度250℃,检测器温度280℃,柱箱初始温度50℃,以10℃
·
min
‑1升温至250℃,保持5min;2
‑
乙酰基吡嗪的收率为30.5%,有少量乙酰基二取代副产物生成。
[0049]
实施例3
[0050]
采用h型电解槽作为反应器,阳极镀钌钛板,阴极铅板,阳极室和阴极室用阳离子交换膜nafion pfsa membranes(n
‑
324)隔开。
[0051]
用二氯甲烷溶解0.067mol吡嗪、0.033mol丙酮酸定容至100ml,取100ml饱和过流酸铵溶液,将上述溶液加入阴极室中,再添加5g二甲基亚砜。
[0052]
所述吡嗪和丙酮酸物质的量比为2:1,所述丙酮酸溶于二氯甲烷的浓度为0.33mol
·
l
‑1,氧化剂过硫酸铵溶于水的浓度为1.5mol
·
l
‑1,所述二甲基亚砜的质量为阴极室中吡嗪/丙酮酸溶液和过硫酸铵溶液总质量的2%;
[0053]
阳极室中加入200ml质量分数20%的硫酸溶液;保持恒温25℃搅拌,通电电解,保持电流密度为800a
·
m
‑2,通电量达到2f
·
mol
‑1后停止通电,分出有机相,得到含有2
‑
乙酰基吡嗪的二氯甲烷溶液,2
‑
乙酰基吡嗪收率以吡嗪计;取样分析,采用上海天美公司gc
‑
7900型气相色谱仪,美国安捷伦公司tm
‑
5毛细管柱进行分析,色谱升温程序为:进样口温度250℃,检测器温度280℃,柱箱初始温度50℃,以10℃
·
min
‑1升温至250℃,保持5min;2
‑
乙酰基吡嗪的收率为24.8%,没有副产物生成。
[0054]
实施例4
[0055]
采用h型电解槽作为反应器,阳极镀钌钛板,阴极铅板,阳极室和阴极室用阳离子交换膜nafion pfsa membranes(n
‑
324)隔开。
[0056]
用二氯甲烷溶解0.067mol吡嗪、0.033mol丙酮酸定容至100ml,取100ml饱和过流酸铵溶液,将上述溶液加入阴极室中,再添加12.5g二甲基亚砜。
[0057]
所述吡嗪和丙酮酸物质的量比为2:1,所述丙酮酸溶于二氯甲烷的浓度为0.33mol
·
l
‑1,氧化剂过硫酸铵溶于水的浓度为1.5mol
·
l
‑1,所述二甲基亚砜的质量为阴极室中吡嗪/丙酮酸溶液和过硫酸铵溶液总质量的5%;
[0058]
阳极室中加入200ml质量分数20%的硫酸溶液;保持恒温25℃搅拌,通电电解,保持电流密度为100a
·
m
‑2,通电量达到2f
·
mol
‑1后停止通电,分出有机相,得到含有2
‑
乙酰基吡嗪的二氯甲烷溶液,2
‑
乙酰基吡嗪收率以吡嗪计;取样分析,采用上海天美公司gc
‑
7900型气相色谱仪,美国安捷伦公司tm
‑
5毛细管柱进行分析,色谱升温程序为:进样口温度
250℃,检测器温度280℃,柱箱初始温度50℃,以10℃
·
min
‑1升温至250℃,保持5min;2
‑
乙酰基吡嗪的收率为37.8%,无副产物生成。
[0059]
实施例5
[0060]
该实施例是水相重复利用的实施例
[0061]
采用h型电解槽作为反应器,阳极镀钌钛板,阴极铅板,阳极室和阴极室用阳离子交换膜nafion pfsa membranes(n
‑
324)隔开。
[0062]
取实施例1中的阴极水相溶液,加入15g过硫酸铵,配置成饱和过硫酸铵溶液;用二氯甲烷溶解0.033mol吡嗪、0.033mol丙酮酸定容至100ml;将上述溶液加入阴极室中,再添加5g二甲基亚砜。
[0063]
所述阴极水相溶液是前一次反应结束通过分液得到的水溶液,所述丙酮酸溶于二氯甲烷的浓度为0.33mol
·
l
‑1,氧化剂过硫酸铵溶于水的浓度为1.5mol
·
l
‑1,所述二甲基亚砜的质量为阴极室中吡嗪/丙酮酸溶液和过硫酸铵溶液总质量的2%;
[0064]
阳极室中加入200ml质量分数20%的硫酸溶液;保持恒温25℃搅拌,通电电解,保持电流密度为100a
·
m
‑2,通电量达到2f
·
mol
‑1后停止通电,分出有机相,得到含有2
‑
乙酰基吡嗪的二氯甲烷溶液,2
‑
乙酰基吡嗪收率以吡嗪计;取样分析,采用上海天美公司gc
‑
7900型气相色谱仪,美国安捷伦公司tm
‑
5毛细管柱进行分析,色谱升温程序为:进样口温度250℃,检测器温度280℃,柱箱初始温度50℃,以10℃
·
min
‑1升温至250℃,保持5min;2
‑
乙酰基吡嗪的收率为40.0%,无副产物生成,可实现水相的重复利用。
[0065]
对比例1
[0066]
在1000ml三口烧瓶,加入镁屑9g,无水四氢呋喃180g,在通入氯甲烷制备甲基氯化镁;加入400g无水甲苯,蒸馏回收四氢呋喃;在0~50℃的条件下滴加20g氰基吡嗪和50g无水甲苯的混合液,保温2~3h;用乙酸中和ph至5~6,滴加150g水进行水解,得到乙酰基吡嗪;用甲苯将乙酰基吡嗪完全萃取,脱除溶剂后得到2
‑
乙酰基吡嗪12g,收率约50%。该技术需先制备格氏试剂,再经反应、水解得到2
‑
乙酰基吡嗪,步骤较多,用到大量有机物,成本较高,且制备格氏试剂危险性大,要求高设备。而电解法一步即可得到2
‑
乙酰基吡嗪,仅用到少量的二氯甲烷为溶剂,使用价格低廉的过硫酸铵为氧化剂,安全环保,简单易行。
[0067]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。