:
1.本发明涉及前列腺素的制造方法。
背景技术:
2.前列腺素(pg)为花生四烯酸在生物体内接受环氧合酶所引起的代谢而合成的内源性生理活性物质群的总称。前列腺素有很多种类,已知有前列腺素h2、前列腺素d2、前列腺素e1、前列腺素e2、前列腺素f
2α
、前列腺素i2等。前列腺素介由各自特异性的g蛋白质偶联型受体参与多种生理功能。
3.前列腺素的化学结构为具备具有4个不对称碳的环戊烷环和2个脂肪族侧链的特征性结构。因此,一直以来,作为合成研究的靶标或者药物开发的起点而受到关注,迄今为止已经开发出各种各样的前列腺素衍生物。被用作其共同中间体的(3as,4r,5s,6ar)
‑
( )
‑
六氢
‑5‑
羟基
‑4‑
(羟甲基)
‑
2h
‑
环戊并[b]呋喃
‑2‑
酮也被称为“科立(corey)内酯”。将科立内酯和代表性的市售的前列腺素衍生物的化学结构示于以下。
[0004][0005]
在科立内酯被开发出来后,前列腺素衍生物的合成中的主要课题为存在于侧链的取代基的立体化学的控制。特别是为了导入具有期望的立体化学的羟基,进一步进行了大量的研究。专利文献1报道了:将相应的羰基还原后利用柱色谱将非对映异构体分离的方法;对通过还原而生成的羟基导入进一步的取代基,通过结晶化等来提高光学纯度的方法。专利文献2公开了通过偶联反应使具有期望的立体化学的侧链单元与环戊烷衍生物结合的方法。专利文献3研究了将相应的羰基用(
‑
)
‑
dip
‑
氯化物
tm
等光学活性的还原剂还原的技术。
[0006]
现有技术文献
[0007]
专利文献
[0008]
专利文献1:国际公开第2012/011128号
[0009]
专利文献2:美国专利申请公开第2007/167641号说明书
[0010]
专利文献3:中国专利申请公开第105985371号
[0011]
专利文献4:欧州专利申请公开第2837621号
[0012]
专利文献5:国际公开第2002/096898号
[0013]
专利文献6:国际公开第2007/091697号
[0014]
专利文献7:美国专利第6248783号说明书
[0015]
专利文献8:美国专利第3726983号说明书
[0016]
专利文献9:美国专利申请公开第2009/259066号说明书
[0017]
非专利文献
[0018]
非专利文献1:e.j.corey et al.,j.am.chem.soc.,1969,91,5675.
技术实现要素:
[0019]
发明所要解决的问题
[0020]
但是,就上述方法而言,利用柱色谱的纯化、衍生物化所产生的步骤数的增加、化学计量的光学活性试剂的使用等成为必须的,经济上的负荷和环境负荷令人担忧。因此,本发明的目的在于,提供使用光学活性催化剂的前列腺素的高效制造方法及其合成中间体。
[0021]
用于解决问题的方法
[0022]
本发明提供以下的[1]~[6]。
[0023]
[1]一种式(1a)、(1b)或(1c)所示的化合物的制造方法,
[0024][0025]
[式中,r1为被苯基取代或未取代的c1‑8烷基,r2为式(2a)、(2b)或(2c)所示的基团(式中,r3为羟基、c1‑3烷氧基、单c1‑3烷基氨基或二c1‑3烷基氨基。)。]
[0026][0027]
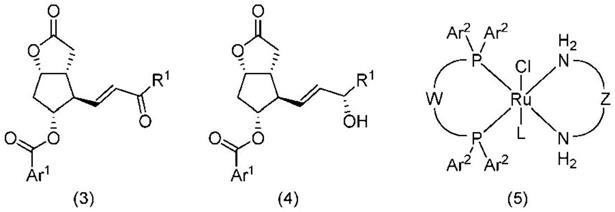
所述制造方法包括在式(5)所示的金属络合物、无机碱和溶剂存在下在氢气气氛下将式(3)所示的化合物还原而得到式(4)所示的化合物的步骤。
[0028][0029]
[式中,ar1为取代或未取代的芳基,
[0030]
各ar2各自独立地为苯基、3,5
‑
二甲基苯基或3,5
‑
二(叔丁基)
‑4‑
甲氧基苯基,
[0031]
w为取代或未取代的联苯基或者取代或未取代的联萘基,
[0032]
z为被选自苯基、c1‑3烷氧基苯基和c1‑8烷基中的2个以上基团取代的亚乙基,l为氯原子或者在z具有苯基或c1‑3烷氧基苯基时为构成上述苯基或上述c1‑3烷氧基苯基的碳原子之一。]
[0033]
[2]根据[1]所述的方法,其中,溶剂包含选自醚、醇、乙腈和水中的至少1种溶剂。
[0034]
[3]根据[1]或[2]所述的方法,其中,ar1为苯基,ar2为3,5
‑
二甲基苯基。
[0035]
[4]根据[1]~[3]中任一项所述的方法,其中,r1为正戊基、2
‑
甲基己基或2
‑
苯基乙基。
[0036]
[5]根据[1]~[4]中任一项所述的方法,其中,还包括通过式(4)所示的化合物与式(6)所示的化合物的反应得到式(7)所示的化合物的步骤。
[0037][0038]
[式中,r1为被苯基取代或未取代的c1‑8烷基,ar1和ar3各自独立地为芳基,x为离去基团。]
[0039]
[6]式(7’)所示的化合物。
[0040][0041]
[式中,r为被苯基取代或未取代的c1‑8烷基,a为硝基。]
[0042]
发明效果
[0043]
根据本发明,通过进行使用光学活性催化剂的催化加氢还原,能够以高的立体选择性导入侧链的羟基,能够高效地制造前列腺素衍生物。
具体实施方式
[0044]
以下对本发明的一个实施方式进行详述。本说明书中,为了方便,也将“式(1)所示的化合物”等称为“化合物(1)”等。
[0045]
本发明的一个实施方式为化合物(1a)、(1b)或(1c)的制造方法。
[0046][0047]
式(1a)、(1b)或(1c)中,r1为未取代的或被苯基取代的c1‑8烷基。c1‑8烷基是碳原子数为1~8个的直链状或支链状烷基,也可以为光学活性的支链状烷基。作为c1‑8烷基,可列举例如甲基、乙基、1
‑
丙基、2
‑
丙基(也称为异丙基)、1
‑
丁基、2
‑
甲基
‑1‑
丙基(也称为异丁
基)、1
‑
甲基
‑2‑
丙基、叔丁基、1
‑
戊基、2
‑
甲基
‑1‑
丁基、3
‑
戊基、3
‑
甲基
‑1‑
丁基(也称为异戊基)、2,2
‑
二甲基
‑1‑
丙基(也称为新戊基)、1
‑
己基、1
‑
甲基
‑2‑
戊基、2
‑
甲基
‑2‑
戊基、3
‑
甲基
‑2‑
戊基、1
‑
庚基、2
‑
庚基、2
‑
甲基
‑1‑
己基、2
‑
乙基
‑1‑
戊基、3
‑
甲基
‑1‑
己基、3
‑
乙基
‑1‑
戊基、3
‑
庚基、4
‑
庚基、1
‑
辛基、2
‑
辛基、2
‑
甲基
‑1‑
庚基、3
‑
乙基
‑1‑
戊基、3
‑
丙基
‑1‑
丁基等。另外,被苯基取代的c1‑8烷基是构成上述碳原子数为1~8的直链状或支链状烷基的氢原子的一部分被取代为苯基后的基团。作为被苯基取代的c1‑8烷基的具体例,可列举苯基甲基(也称为苄基)、1
‑
苯基乙基、2
‑
苯基乙基、1
‑
苯基丙基、1
‑
甲基
‑1‑
苯基乙基等。
[0048]
式(1a)、(1b)或(1c)中,r2为式(2a)、(2b)或(2c)所示的基团,r3为羟基、c1‑3烷氧基、单c1‑3烷基氨基或二c1‑3烷基氨基。
[0049][0050]
c1‑3烷氧基为键合有碳原子数为1~3个的直链状或支链状烷基的氧原子所形成的基团。作为c1‑3烷氧基,可列举例如甲氧基、乙氧基、1
‑
丙氧基、2
‑
丙氧基(也称为异丙氧基)。
[0051]
单c1‑3烷基氨基为键合有1个碳原子数为1~3个的直链状或支链状烷基的氨基所形成的基团。作为单c1‑3烷基氨基,可列举例如单甲基氨基、单乙基氨基、单1
‑
丙基氨基、单2
‑
丙基氨基。
[0052]
二c1‑3烷基氨基为键合有2个碳原子数为1~3个的直链状或支链状烷基的氨基所形成的基团。作为二c1‑3烷基氨基,可列举例如n,n
‑
二甲基氨基、n
‑
乙基
‑
n
‑
甲基氨基、n,n
‑
二乙基氨基、n
‑
乙基
‑
n
‑
(1
‑
丙基)氨基、n,n
‑
二(1
‑
丙基)氨基、n,n
‑
二(2
‑
丙基)氨基。
[0053]
(步骤1)
[0054]
本实施方式的方法包括在金属络合物(5)、无机碱和溶剂存在下在氢气气氛下将化合物(3)还原而得到化合物(4)的步骤。式(3)或(4)中,r1与式(1)中的定义相同,ar1为取代或未取代的芳基。
[0055][0056]
芳基为芳香族烃基,例如,可以为碳原子数6~10的芳香族烃基。作为芳基,可列举例如苯基、萘基。芳基可被卤素原子(例如氟原子、氯原子、溴原子、碘原子)、c1‑3烷基、c1‑3烷氧基或氰基取代。
[0057]
式(5)中,各ar2各自独立地为苯基、3,5
‑
二甲基苯基或3,5
‑
二(叔丁基)
‑4‑
甲氧基苯基。优选4个ar2为彼此相同的基团。
[0058]
式(5)中,w为取代或未取代的联苯基或者取代或未取代的联萘基。另外,(ar2)2p
‑
w
‑
p(ar2)2为能够与钌原子配位的双齿配体,w优选为作为单一的阻转异构体的双(亚甲基二氧基苯)基或联萘基。作为(ar2)2p
‑
w
‑
p(ar2)2,可列举例如2,2
’‑
双(二苯基膦基)
‑
1,1
’‑
联萘(binap)、2,2
’‑
双[二(对甲苯基)膦基]
‑
1,1
’‑
联萘(tolbinap)、2,2
’‑
双[二(间甲苯基)膦基]
‑
1,1
’‑
联萘、2,2
’‑
双[二(3,5
‑
二甲苯基)膦基]
‑
1,1
’‑
联萘(xylyl
‑
binap)、2,2
’‑
双[二(对叔丁基苯基)膦基]
‑
1,1
’‑
联萘、2,2
’‑
双[二(对甲氧基苯基)膦基]
‑
1,1
’‑
联萘、2,2
’‑
双[二(3,5
‑
二叔丁基
‑4‑
甲氧基苯基)膦基]
‑
1,1
’‑
联萘、2,2
’‑
双[二(环戊基)膦基]
‑
1,1
’‑
联萘、2,2
’‑
双[二(环己基)膦基]
‑
1,1
’‑
联萘、2,2
’‑
双(二苯基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘、2,2
’‑
双(二对甲苯基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘、2,2
’‑
双(二间甲苯基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘、2,2
’‑
双(二3,5
‑
二甲苯基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘(xylyl
‑
h8
‑
binap)、2,2
’‑
双(二对叔丁基苯基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘、2,2
’‑
双(二对甲氧基苯基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘、2,2
’‑
双(二对氯苯基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘、2,2
’‑
双(二环戊基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘、2,2
’‑
双(二环己基膦基)
‑
5,5’,6,6’,7,7’,8,8
’‑
八氢
‑
1,1
’‑
联萘、((4,4
’‑
联
‑
1,3
‑
苯并二氧杂环戊烯)
‑
5,5
’‑
二基)双(二苯基膦)(segphos)、(4,4
’‑
联
‑
1,3
‑
苯并二氧杂环戊烯)
‑
5,5
’‑
二基)双(二(3,5
‑
二甲苯基)膦)(dm
‑
segphos)、((4,4
’‑
联
‑
1,3
‑
苯并二氧杂环戊烯)
‑
5,5
’‑
二基)双(二(3,5
‑
二叔丁基
‑4‑
甲氧基苯基)膦)、((4,4
’‑
联
‑
1,3
‑
苯并二氧杂环戊烯)
‑
5,5
’‑
二基)双(二(4
‑
甲氧基苯基)膦)、((4,4
’‑
联
‑
1,3
‑
苯并二氧杂环戊烯)
‑
5,5
’‑
二基)双(二环己基膦)、((4,4
’‑
联
‑
1,3
‑
苯并二氧杂环戊烯)
‑
5,5
’‑
二基)双(双(3,5
‑
二叔丁基苯基)膦)、2,2
’‑
双(二
‑
3,5
‑
二甲苯基膦基)
‑
6,6
’‑
二甲氧基
‑
1,1
’‑
联苯(xylyl
‑
meo
‑
biphep)、2,2
’‑
双(二苯基膦基)
‑
6,6
’‑
二甲基
‑
1,1
‑
联苯2,2
’‑
双(二对甲苯基膦基)
‑
6,6
’‑
二甲基
‑
1,1
’‑
联苯、2,2
’‑
双(二邻甲苯基膦基)
‑
6,6
’‑
二甲基
‑
1,1
’‑
联苯、2,2
’‑
双(二间氟苯基膦基)
‑
6,6
’‑
二甲基
‑
1,1
’‑
联苯、2,2
’‑
双(二苯基膦基)
‑
6,6
’‑
二甲氧基
‑
1,1
’‑
联苯、2,2
’‑
双(二对甲苯基膦基)
‑
6,6
’‑
二甲氧基
‑
1,1
’‑
联苯、2,2’,6,6
’‑
四甲氧基
‑
4,4
’‑
双(二
‑
3,5
‑
二甲苯基膦基)
‑
3,3
’‑
联吡啶(xylyl
‑
p
‑
phos)、2,2’,6,6
’‑
四甲氧基
‑
4,4
’‑
双(二苯基膦基)
‑
3,3
’‑
联吡啶、2,2’,6,6
’‑
四甲氧基
‑
4,4
’‑
双(二对甲苯基膦基)
‑
3,3
’‑
联吡啶、2,2’,6,6
’‑
四甲氧基
‑
4,4
’‑
双(二邻甲苯基膦基)
‑
3,3
’‑
联吡啶、4,12
‑
双(二
‑
3,5
‑
二甲苯基膦基)
‑
[2.2]
‑
对环芳烷、4,12
‑
双(二苯基膦基)
‑
[2.2]
‑
对环芳烷、4,12
‑
双(二对甲苯基膦基)
‑
[2.2]
‑
对环芳烷、4,12
‑
双(二邻甲苯基膦基)
‑
[2.2]
‑
对环芳烷、1,1
’‑
双(2,4
‑
二乙基膦)二茂铁、1,13
‑
双(二苯基膦基)
‑
7,8
‑
二氢
‑
6h
‑
二苯并[f,h][1,5]二氧杂环壬四烯、1,13
‑
双(双(3,5
‑
二甲基苯基)膦基)
‑
7,8
‑
二氢
‑
6h
‑
二苯并[f,h][1,5]二氧杂环壬四烯(xylyl
‑
c3
‑
tunephos)、6,6
’‑
双(双(3,5
‑
二甲基苯基)膦基)
‑
2,2’,3,3
’‑
四氢
‑
5,5
’‑
联
‑
1,4
‑
苯并二氧杂环己烯(xylyl
‑
synphos)等。
[0059]
式(5)中,z可以为被选自苯基、c1‑3烷氧基苯基和c1‑8烷基中的2个以上基团取代的亚乙基,这种情况下,l为氯原子。h2n
‑
z
‑
nh2为能够与钌原子配位的双齿配体。作为h2n
‑
z
‑
nh2,可列举例如(s,r)
‑
1,2
‑
二苯基乙二胺、(r,s)
‑
1,2
‑
二苯基乙二胺、1,2
‑
双(4
‑
甲氧基苯基)乙二胺、1
‑
甲基
‑
2,2
‑
二苯基乙二胺、1
‑
异丁基
‑
2,2
‑
二苯基乙二胺、1
‑
异丙基
‑
2,2
‑
二苯基乙二胺(dpipen)、1
‑
甲基
‑
2,2
‑
双(4
‑
甲氧基苯基)乙二胺(damen)、1
‑
异丁基
‑
2,2
‑
双(4
‑
甲氧基苯基)乙二胺、1
‑
异丙基
‑
2,2
‑
双(4
‑
甲氧基苯基)乙二胺(daipen)、1
‑
苯基
‑
2,2
‑
双
(4
‑
甲氧基苯基)乙二胺、1,1
‑
双(4
‑
甲氧基苯基)乙二胺(daen)、1
‑
异丙基
‑
2,2
‑
双(3
‑
甲氧基苯基)乙二胺(3
‑
daipen)等。优选的h2n
‑
z
‑
nh2为光学活性的1,2
‑
二苯基乙二胺或1
‑
异丙基
‑
2,2
‑
二(4
‑
甲氧基苯基)乙二胺。
[0060]
另外,在z具有苯基或c1‑3烷氧基苯基时,构成该苯基或c1‑3烷氧基苯基的碳原子之一可以与钌原子直接结合而形成钌杂双环[2.2.1]庚烷结构。
[0061]
步骤1中,在规定的容器中使化合物(3)溶解于溶剂,加入无机碱,用不活泼气体置换容器的气相。然后,在加入金属络合物(5)之后,用氢气置换气相,在氢气气氛下搅拌规定的时间。为了使气相中的氢气容易混合到反应溶液中,优选进行剧烈搅拌。
[0062]
溶剂为能够溶解化合物(3)且不抑制催化加氢还原的溶剂即可。溶剂可列举:苯、甲苯、二甲苯等芳香族烃;己烷、庚烷等脂肪族烃;二氯甲烷、1,2
‑
二氯乙烷、氯苯、二氯苯等卤代烃;乙醚、二异丙基醚、四氢呋喃、甲基叔丁基醚、环戊基甲基醚、2
‑
甲基四氢呋喃等醚;甲醇、乙醇、异丙醇、正丁醇、2
‑
丁醇、叔丁醇、乙二醇、1,3
‑
丙二醇、1,2
‑
丙二醇、甘油等醇;乙腈、n,n
‑
二甲基甲酰胺(dmf)、n,n
‑
二甲基乙酰胺等。这些溶剂可以以单一溶剂形式使用,也可以任选进行混合而使用。只要能够溶解化合物(3),则也可以混合水。优选的溶剂为选自醚、醇、乙腈和水中的至少1种。
[0063]
溶剂的使用量优选为使化合物(3)的摩尔浓度达到0.05~1.5摩尔/l的量,更优选为达到0.1~0.9摩尔/l的量。
[0064]
无机碱可以为碱性的无机盐。作为无机碱,可列举例如碳酸钾、碳酸钠、碳酸铯、碳酸氢钾、碳酸氢钠、氢氧化钾、氢氧化钠、磷酸三钾、磷酸三钠、磷酸氢二钾、磷酸氢二钠、磷酸二氢钾、磷酸二氢钠。
[0065]
无机碱的使用量可以是相对于金属络合物(5)的摩尔数为0.1~50当量,更优选为1~20当量。
[0066]
金属络合物(5)的使用量根据化合物(3)的量、反应条件和金属络合物(5)的种类而不同,通常相对于化合物(3)的摩尔数为0.1~25摩尔%,更优选为1~5摩尔%。
[0067]
反应温度根据所使用的溶剂的种类而不同,可以在
‑
50~50℃下进行。优选的反应温度为
‑
20~10℃。若反应温度过低,则化合物(3)的溶解性有时会下降,若反应温度过高,则化合物(3)、化合物(4)或金属络合物(5)有时会分解。
[0068]
反应可以在常压下进行,也可以在规定的压力下进行。例如,可以通过向橡胶制的气球中封入氢气、并且将气球连接到容器(烧瓶等)的开口部的方法来进行。由于氢气会随着反应的进行而被消耗,因此使用足够量的氢气或在反应过程中补充氢气。
[0069]
反应结束时,将容器的气相用不活泼气体置换,然后,可以根据需要滤除金属络合物并单独或适当组合使用萃取、结晶化、蒸馏、各种色谱法等通常使用的纯化方法进行纯化。
[0070]
(步骤2)
[0071]
本实施方式的方法可以还包括通过化合物(4)与化合物(6)的反应得到化合物(7)的步骤。式(7)中,r1和ar1与式(4)中的定义相同。
[0072][0073]
式(6)或式(7)中,ar3为取代或未取代的芳基。ar3中的取代或未取代的芳基可以与ar1中的定义相同,但是优选ar1与ar3为彼此不同的基团。优选的ar3为邻硝基苯基、间硝基苯基或对硝基苯基。
[0074]
化合物(6)为将化合物(4)的羟基酰基化的试剂。式(6)中,x为离去基团。作为离去基团,可列举例如卤素原子(例如氟原子、氯原子、溴原子)、体积较大的酰氧基(例如特戊酰氧基)。
[0075]
步骤2中,例如,在规定的容器中向化合物(4)中加入根据需要使用的溶剂、碱和添加剂,加入化合物(6)后搅拌规定的时间。
[0076]
反应可以在无溶剂下进行,也可以在溶剂中进行。反应优选在有机溶剂中进行。所使用的溶剂只要为能够溶解化合物(4)、化合物(6)且不抑制反应的溶剂即可。作为溶剂,可列举例如:苯、甲苯、二甲苯等芳香族烃;己烷、庚烷等脂肪族烃;乙酸乙酯、乙酸异丙酯等羧酸酯;二氯甲烷、1,2
‑
二氯乙烷、氯苯、二氯苯等卤代烃;乙醚、二异丙基醚、四氢呋喃、甲基叔丁基醚、环戊基甲基醚、2
‑
甲基四氢呋喃等醚。这些溶剂可以以单一溶剂形式使用,也可以任意地进行混合而使用。溶剂的使用量优选为使化合物(4)的摩尔浓度达到0.2~1.2摩尔/l的量.
[0077]
为了加快反应,也可以向反应溶液中加入碱。作为碱,可列举例如:三乙基胺、n,n
‑
二乙基
‑
n
‑
异丙基胺等三烷基胺;吡啶、2,6
‑
二甲基吡啶等含氮芳香族化合物。碱的使用量根据化合物(4)的量而不同,优选相对于化合物(4)的摩尔数为1~10当量。
[0078]
另外,为了进一步加快反应,还可以加入4
‑
二甲基氨基吡啶、咪唑、n
‑
甲基咪唑等添加剂。添加剂的使用量根据化合物(4)的量以及碱的种类和使用量而不同,优选相对于化合物(4)的摩尔数为0.001~0.05当量。
[0079]
反应可以在通常适合于酰基化反应的温度下进行。反应温度可以为例如
‑
20~60℃,优选为0~30℃。
[0080]
对于生成物,可以单独或适当组合使用萃取、结晶化、蒸馏、各种色谱法等通常可使用的纯化法来进行纯化。
[0081]
(步骤3)
[0082]
本实施方式的方法可以还包括通过在溶剂中用碱进行处理而将化合物(7)转化为化合物(8)的步骤。式(8)中,r1与式(7)中的定义相同。
[0083][0084]
步骤3中,例如,在规定的容器中向化合物(7)中加入溶剂并进行混合,加入碱后搅拌规定的时间。
[0085]
步骤3中的碱可以为有机碱,也可以为无机碱。作为有机碱,可列举例如:三乙基胺、n,n
‑
二乙基
‑
n
‑
异丙基胺等三烷基胺;吡啶、2,6
‑
二甲基吡啶等含氮芳香族化合物;甲醇钠、甲醇钾、乙醇钠、乙醇钾、叔丁醇钠、叔丁醇钾等金属醇盐。作为无机碱,可列举例如碳酸钾、碳酸钠、碳酸铯、碳酸氢钾、碳酸氢钠、氢氧化钾、氢氧化钠、磷酸三钾、磷酸三钠、磷酸氢二钾、磷酸氢二钠、磷酸二氢钾、磷酸二氢钠。碱的使用量根据化合物(7)的量而不同,优选相对于化合物(7)的摩尔数为1~10当量。
[0086]
步骤3中的溶剂可列举例如:苯、甲苯、二甲苯等芳香族烃;己烷、庚烷等脂肪族烃;二氯甲烷、1,2
‑
二氯乙烷、氯苯、二氯苯等卤代烃;乙醚、二异丙基醚、四氢呋喃、甲基叔丁基醚、环戊基甲基醚、2
‑
甲基四氢呋喃等醚;甲醇、乙醇、异丙醇、正丁醇、2
‑
丁醇、叔丁醇、乙二醇、1,3
‑
丙二醇、1,2
‑
丙二醇、甘油等醇;乙腈、n,n
‑
二甲基甲酰胺(dmf)、n,n
‑
二甲基乙酰胺等。这些溶剂可以以单一溶剂形式使用,也可以任意地进行混合而使用。优选包含水或醇。
[0087]
溶剂的使用量优选为使化合物(7)的摩尔浓度达到0.05~1.0摩尔/l的量,更优选为达到0.1~0.5摩尔/l的量。
[0088]
对于反应生成物,可以单独或适当组合使用萃取、结晶化、蒸馏、各种色谱法等通常可使用的纯化方法来进行纯化。
[0089]
本实施方式的方法可以还包括用于从化合物(8)转化为化合物(1a)或(1b)的步骤。这些步骤可以参考专利文献1~4和非专利文献1的记载。
[0090]
(步骤4)
[0091]
步骤4为用于得到化合物(1a)的步骤,可以包括两个以上阶段。化合物(1a)可以是例如地诺前列素等前列腺素f
2α
衍生物、比马前列素等。
[0092]
(步骤5)
[0093]
步骤5为用于得到化合物(1b)的步骤,可以包括两个以上阶段。化合物(1b)可以是例如前列地尔、利马前列素等前列腺素e1衍生物、地诺前列酮等前列腺素e2衍生物。
[0094]
(步骤6)
[0095]
步骤6为用于得到化合物(1c)的步骤,可以包括两个以上阶段。化合物(1c)可以为例如拉坦前列素等前列腺素f
2α
衍生物。
[0096]
本发明的另一个实施方式为化合物(7’)。
[0097][0098]
式(7’)中,r为未取代或被苯基取代的c1‑8烷基。r的定义与式(1a)中的r1的定义相同。
[0099]
式(7’)中,a为键合于苯环的邻位、间位、对位中的任一位置的硝基。
[0100]
化合物(7’)可以为单一的非对映异构体,也可以为两种以上非对映异构体的混合物。为了制造化合物(1a)、(1b)或(1c),优选化合物(7’)的光学纯度高。
[0101]
化合物(7’)的光学纯度可以通过进行重结晶来提高。作为结晶化中使用的溶剂,可列举例如:苯、甲苯、二甲苯等芳香族烃;己烷、庚烷等脂肪族烃;乙酸乙酯、乙酸异丙酯等羧酸酯;二氯甲烷、1,2
‑
二氯乙烷、氯苯、二氯苯等卤代烃;乙醚、二异丙基醚、四氢呋喃、甲基叔丁基醚、环戊基甲基醚、2
‑
甲基四氢呋喃等醚;甲醇、乙醇、异丙醇、1
‑
丁醇等醇等。这些溶剂可以以单一溶剂形式使用,也可以任意地进行混合而使用。优选包含羧酸酯或醇。通过将光学纯度低的化合物(7’)根据需要进行加热而溶解于溶剂并缓慢地冷却,能够得到光学纯度更高的化合物(7’)。
[0102]
化合物(7’)的结晶性优良,作为用于制造光学纯度高的化合物(1a)、(1b)或(1c)的制造中间体有用。
[0103]
实施例
[0104]
使用实施例更详细地说明本发明的一个实施方式。
[0105]
以下的说明中使用的缩写通常应按照该领域的技术常识来理解。缩写的含义具体如下。
[0106]
bz:苯甲酰基
[0107]
can:硝酸铈(iv)铵
[0108]
csa:10
‑
樟脑磺酸
[0109]
dibal
‑
h:二异丁基氢化铝
[0110]
dmap:4
‑
二甲基氨基吡啶
[0111]
dmf:n,n
‑
二甲基甲酰胺
[0112]
et:乙基
[0113]
ipa:异丙醇
[0114]
n
‑
:正
[0115]
nmr:核磁共振谱
[0116]
p
‑
:对
[0117]
ph:苯基
[0118]
pmb:对甲氧基苄基
[0119]
me
‑
thf:2
‑
甲基四氢呋喃
[0120]
tea:三乙胺
[0121]
tert:叔
[0122]
thf:四氢呋喃
[0123]
thp:四氢吡喃
‑2‑
基
[0124]1h
‑
nmr谱用以四甲基硅烷作为内标(0ppm)校正后的化学位移值(δ)来表示,裂分图案以下述方式来缩写。s:单峰、d:双峰、t:三重峰、q:四重峰、dd:双二重峰、ddd:双二倍二重峰、dt:双三重峰、m:多重峰、br:宽峰。
[0125]
实施例1:地诺前列酮的制造方法
[0126][0127]
(步骤1)
[0128]
通过专利文献5的实施例1和专利文献6的实施例8中记载的方法,由市售的(
‑
)
‑
corey lactone
‑
benzoate(商品名)和2
‑
氧代
‑
庚基膦酸二甲酯制备化合物(3a)。将化合物(3a)(20.0g、54mmol)和磷酸三钾(1.1g)悬浮于thf(100ml)与乙醇(20ml)的混合液中。在冰浴下用氮气置换容器内的气相。向悬浮液中加入(r)
‑
rucy
‑
xylbinap(东京化成工业、0.64g、0.52mmol)后,用氢气置换气相,搅拌24小时。用氮气置换气相后,减压馏去溶剂,得到化合物(4a)(非对映异构体过量率87%)。得到的化合物(4a)不进行进一步的纯化而直接用于步骤2。
[0129]
(步骤2)
[0130]
向化合物(4a)中加入乙酸乙酯(50ml)而将其溶解,加入tea(10.9g)和dmap(33mg)。向得到的溶液中滴加对硝基苯甲酰氯(14.0g)的乙酸乙酯溶液(40ml),搅拌1小时。向反应溶液中加入乙酸乙酯,用水洗涤,回收有机层。减压馏去溶剂后,向得到的残渣中加入乙醇(100ml),加热使其溶解,并逐渐冷却到0℃而得到晶体。用乙醇洗涤所得到的晶体,在减压下进行干燥,得到化合物(7a)(21.1g、40mmol、收率74%、非对映异构体过量率99%)。
[0131]
mp:105℃;
[0132]1hnmr(400mhz,cdcl3):δ8.26
‑
8.21(m,2h),8.14
‑
8.09(m,2h),7.91(dd,2h),7.53(dd,1h),7.38(dd,2h),5.76
‑
5.65(m,2h),5.46(ddd,1h),5.25(ddd,1h),5.06(ddd,1h),2.90
‑
2.74(m,3h),2.61(ddd,1h),2.51(d,1h),2.22(ddd,1h),1.74(m,2h),1.36
‑
1.26(m,6h),0.86(t,3h);
[0133]
13
cnmr(100mhz,cdcl3):δ176.1,165.8,163.8,150.4,135.7,133.3,132.0,131.3,130.6,129.5,129.4,128.4,123.5,83.0,78.5,75.8,53.9,42.4,37.5,34.7,34.2,31.4,24.8,22.4,13.9
[0134]
(步骤3)
[0135]
将化合物(7a)(20.0g、38mmol)的甲醇溶液(100ml)用冰冷却,加入碳酸钾(10.6g),反应4小时。加入用水稀释后的磷酸,用甲苯/己烷混合液(体积比1/1)进行洗涤。减压馏去溶剂,将得到的残渣用乙酸乙酯萃取,回收有机层。将有机层的溶剂减压馏去,得到化合物(8a)(9.8g、37mmol、收率95%)。
[0136]1hnmr(400mhz,cdcl3):δ5.60(dd,1h),5.44(dd,1h),4.88(ddd,1h),4.08
‑
4.03(m,1h),3.97
‑
3.91(m,1h),2.78
‑
2.69(m,1h),2.62
‑
2.49(m,2h)2.47
‑
2.38(m,2h),2.31
‑
2.22(m,2h),1.98
‑
1.91(m,1h),1.63
‑
1.42(m,2h),1.38
‑
1.23(m,6h),0.89(t,3h);
[0137]
13
cnmr(100mhz,cdcl3):δ176.9,136.8,130.2,82.4,76.3,72.8,56.12,42.34,39.61,37.05,34.0,31.6,25.1,22.5,13.9)
[0138]
(步骤5)
[0139][0140]
通过专利文献7的实施例14中记载的方法,将化合物(8a)(9.8g、37mmol)转化为化合物(10a)。将化合物(10a)通过专利文献8的实施例1中记载的方法转化为化合物(11a),进一步参考专利文献9的实施例14中记载的方法转化为化合物(9a)(7.2g、19mmol、收率51%(3个步骤))。
[0141]
mp:66℃
[0142]1hnmr(400mhz,cdcl3):δ5.68(dd,1h),5.58(dd,1h),5.47
‑
5.34(m,2h),4.20
‑
4.10(m,1h),4.05(dd,1h),2.75(dd,1h),2.47
‑
1.93(m,9h),1.82
‑
1.43(m,4h),1.40
‑
1.20(m,6h),0.89(t,3h)
[0143]
13
cnmr(100mhz,cdcl3):6214.4,177.6,136.7,130.9,126.9,73.2,72.5,54.6,53.6,46.4,37.2,33.2,31.8,26.4,25.3,24.6,22.8,14.2
[0144]
对于步骤1,在以下的条件下同样地进行催化加氢还原。将反应条件和结果示于表1中。
[0145]
[表1]
[0146][0147]
实施例9:利马前列素的制造方法
[0148][0149]
(步骤1)
[0150]
参考专利文献5和6中记载的方法,由市售的(
‑
)
‑
corey lactone
‑
benzoate(商品名)和2
‑
氧代
‑4‑
甲基辛基膦酸二甲酯制备化合物(3b)。
[0151]1hnmr(300mhz,cdcl3):δ8.02
‑
7.96(m,2h),7.62
‑
7.55(m,1h),7.50
‑
7.42(m,2h),6.68(dd,1h),6.24(d,1h),5.33(q,1h),5.14
‑
5.08(m,1h),2.96
‑
2.85(m,3h),2.68
‑
2.28(m,5h),2.05
‑
1.93(m,1h),1.32
‑
1.15(m,6h),0.92
‑
0.84(m,6h)
[0152]
参考实施例1中记载的方法,由化合物(3b)(20.0g、50mmol)得到化合物(4b)(非对映异构体过量率87%)。
[0153]
(步骤2)
[0154]
参考实施例1中记载的方法,由化合物(4b)得到化合物(7b)(18.1g、33mmol、收率66%、非对映异构体过量率97%)。
[0155]
mp:74℃;
[0156]1hnmr(400mhz,cdcl3):δ8.22
‑
8.19(m,2h),8.10
‑
8.07(m,2h),7.90
‑
7.88(m,2h),7.52
‑
7.49(m,1h),7.37
‑
7.33(m,2h),5.77
‑
5.71(m,1h),5.67
‑
5.61(m,1h),5.53(dd,1h),5.23(dd,1h),5.04(ddd,1h),2.89
‑
2.71(m,3h),2.59(ddd,1h),2.49(d,1h),2.20(ddd,1h),1.64(t,2h),1.49
‑
1.41(m,1h),1.26
‑
1.10(m,6h),0.91(d,3h),0.85(t,3h);
[0157]
13
cnmr(100mhz,cdcl3):δ176.2,165.9,163.9,150.5,135.9,133.4,132.4,131.5,130.7,129.6,128.5,123.6,83.1,78.6,74.7,54.0,42.5,41.5,37.6,36.6,34.8,29.5,29.0,23.0,19.9,14.2
[0158]
(步骤3)
[0159]
参考实施例1中记载的方法,由化合物(7b)(18.1g、33mmol)得到化合物(8b)(9.2g、31mmol、收率95%)。
[0160]1hnmr(400mhz,cdcl3):δ5.56(dd,1h),5.44(dd,1h),4.88(ddd,1h),4.17
‑
4.08(m,1h),3.96
‑
3.89(m,1h),2.78
‑
2.21(m,6h),1.93(ddd,1h),1.87(br,1h),1.63(br,1h),1.45
‑
1.08(m,9h),0.93
‑
0.82(m,6h)
[0161]
(步骤5)
[0162][0163]
(步骤5
‑
1)
[0164]
(3ar,4r,5r,6as)
‑5‑
[(4
‑
甲氧基苄基)氧基]
‑4‑
{(3s,5s,e)
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑5‑
甲基壬
‑1‑
烯
‑1‑
基}六氢
‑
2h
‑
环戊并[b]呋喃
‑2‑
酮(10b)
[0165]
向化合物(8b)(0.29g、0.98mmol)中加入二氯甲烷(3ml)而将其溶解,加入2,2,2
‑
三氯亚氨逐乙酸
‑4‑
甲氧基苄酯(0.69g)和csa(0.01g),在室温下进行搅拌。向反应溶液中加入饱和碳酸氢钠水溶液、乙酸乙酯,用硅藻土进行过滤,减压馏去溶剂。将残渣用超临界液相色谱(装置名:viridis csh fluoro
‑
phenyl obd prep column、二氧化碳/甲醇/氯仿=90/5/5~80/10/10)进行纯化,由此得到化合物(10b)(0.374g、收率71%)。
[0166]1hnmr(400mhz,cdcl3):δ7.25
‑
7.16(m,4h),6.88
‑
6.83(m,4h),5.48(dd,1h),5.41(dd,1h),4.98
‑
4.93(m,1h),4.57
‑
4.21(m,4h),3.81
‑
3.73(m,8h),2.79
‑
2.61(m,3h),2.53
‑
2.46(m,1h),2.34
‑
2.11(m,2h),1.55
‑
1.02(m,7h),0.91
‑
0.82(m,6h)
[0167]
(步骤5
‑
2)
[0168]
(3ar,4r,5r,6as)
‑5‑
[(4
‑
甲氧基苄基)氧基]
‑4‑
{(3s,5s,e)
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑5‑
甲基壬
‑1‑
烯
‑1‑
基}六氢
‑
2h
‑
环戊并[b]呋喃
‑2‑
醇(11b)
[0169]
将化合物(10b)(0.370g、0.69mmol)溶解于二氯甲烷(7ml)中,在
‑
78℃下平稳地滴加dibal
‑
h(1mol/l己烷溶液、0.758ml)。在
‑
78℃下搅拌30分钟后,加入饱和罗谢尔(roscelle)盐水溶液,在室温下搅拌30分钟,加入乙酸乙酯,用硅藻土进行过滤,减压馏去溶剂。将残渣用硅胶柱色谱(庚烷/乙酸乙酯=2/1~1/1)纯化,由此得到化合物(11b)(0.348g、收率94%)。
[0170]1hnmr(400mhz,cdcl3):δ7.25
‑
7.18(m,4h),6.87
‑
6.79(m,4h),5.68
‑
5.36(m,3h),4.68
‑
4.20(m,5h),3.81
‑
3.69(m,8h),2.52
‑
2.17(m,4h),2.06
‑
1.77(m,2h),1.54
‑
1.36(m,3h),1.29
‑
0.99(m,7h),0.91
‑
0.82(m,6h)
[0171]
(步骤5
‑
3)
[0172]
(1s,2r,3r,4r)
‑2‑
[(z)
‑4‑
(1,3
‑
二氧杂环戊烷
‑2‑
基)丁
‑2‑
烯
‑1‑
基]
‑4‑
[(4
‑
甲氧基苄基)氧基]
‑3‑
{(3s,5s,e)
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑5‑
甲基壬
‑1‑
烯
‑1‑
基}环戊醇(13b)
[0173]
将化合物(12b)(2
‑
(1,3
‑
二氧杂环戊烷
‑2‑
基)乙基三苯基溴化鏻、1.40g、3.16mmol)悬浮于thf(6ml)中,加入叔丁醇钾(0.354g、3.16mmol),在室温下搅拌30分钟。在冰冷却下向褐色悬浮状的反应溶液中滴加化合物(11b)(0.340g、0.631mm0l)的thf(2ml)溶液,在0℃下搅拌30分钟。向反应溶液中加入饱和氯化铵水溶液、乙酸乙酯,用硅藻土进行过滤,减压馏去溶剂。将残渣用硅胶柱色谱(庚烷/乙酸乙酯=4/1~2/1、然后氯仿/甲醇=50/1~30/1)纯化,由此以白色固体形态得到化合物(13b)(0.258g、收率66%)。
[0174]1hnmr(400mhz,cdcl3):δ7.25
‑
7.18(m,4h),6.86
‑
6.79(m,4h),5.56
‑
5.40(m,4h),4.93
‑
4.89(m,1h),4.54
‑
4.22(m,4h),4.13
‑
4.07(m,1h),4.01
‑
3.83(m,4h),3.81
‑
3.74(m,8h),3.01
‑
2.97(m,1h),2.68
‑
2.55(m,2h),2.45
‑
2.34(m,2h),2.18
‑
1.99(m,2h),1.90
‑
1.83(m,1h),1.56
‑
1.37(m,4h),1.30
‑
1.01(m,6h),0.89
‑
0.82(m,6h)
[0175]
(步骤5
‑
4)
[0176]
(1s,2r,3r,4r)
‑2‑
[4
‑
(1,3
‑
二氧杂环戊烷
‑2‑
基)丁基]
‑4‑
[(4
‑
甲氧基苄基)氧基]
‑3‑
{(3s,5s,e)
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑5‑
甲基壬
‑1‑
烯
‑1‑
基}环戊醇(14b)
[0177]
将化合物(13b)(0.250g、0.401mmol)溶解于乙酸乙酯(3ml)中,加入rhcl(pph3)3(0.074g、0.080mmol),在氢气气氛下在室温下搅拌5小时。将反应溶液减压浓缩,将得到的残渣用硅胶柱色谱(庚烷/乙酸乙酯=4/1~2/1)、然后氨基硅胶柱色谱(庚烷/乙酸乙酯=4/1~2/1)纯化,由此以白色固体形态得到化合物(14b)(0.208g、收率83%)。
[0178]1hnmr(400mhz,cdcl3):δ7.25
‑
7.18(m,4h),6.87
‑
6.81(m,4h),5.48(dd,1h),5.38(dd,1h),4.86
‑
4.81(m,1h),4.52
‑
4.21(m,4h),4.18
‑
4.12(m,1h),3.98
‑
3.91(m,2h),3.86
‑
3.73(m,10h),2.54
‑
2.47(m,1h),2.20
‑
1.95(m,3h),1.72
‑
1.02(m,18h),0.91
‑
0.81(m,6h)
[0179][0180]
(步骤5
‑
5)
[0181]
(e)
‑7‑
((1r,2r,3r,5s)
‑5‑
羟基
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑2‑
{(3s,5s,e)
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑5‑
甲基壬
‑1‑
烯
‑1‑
基}环戊基)庚
‑2‑
烯酸甲酯(17b)
[0182]
将化合物(14b)(0.190g、0.304mmol)溶解于thf(4ml)中,在冰冷却下加入6mol/l盐酸(2ml),在室温下搅拌过夜。向反应溶液中加入饱和碳酸氢钠水溶液,用乙酸乙酯进行萃取。将有机层用饱和食盐水洗涤,用无水硫酸镁干燥,减压馏去溶剂。将残渣用硅胶柱色谱(庚烷/乙酸乙酯=2/1~1/1)纯化,由此以白色固体形态得到化合物15b的粗纯化物。将得到的粗纯化物溶解于二氯甲烷(4ml)中,在冰冷却下加入(三苯基亚正膦基)乙酸甲酯(16b)(0.508g,1.52mmol),在室温下搅拌2小时。将反应溶液用硅胶柱色谱(庚烷/乙酸乙酯=2/1~1/1)纯化,由此以白色固体形态得到化合物(17b)(0.131g、收率68%)。
[0183]
(步骤5
‑
6)
[0184]
(e)
‑7‑
((1r,2r,3r,5s)
‑5‑
羟基
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑2‑
{(3s,5s,e)
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑5‑
甲基壬
‑1‑
烯
‑1‑
基}环戊基)庚
‑2‑
烯酸(18b)
[0185]
将化合物(17b)(0.0900g、0.141mmol)悬浮于乙醇(2ml)中,在冰冷却下加入1mol/l氢氧化钾水溶液(0.565ml、0.565mmol),在40℃下搅拌3小时。在冰冷却下向反应溶液中加入1mol/l盐酸(0.60ml)、乙酸乙酯,用硅藻土进行过滤,减压馏去溶剂。将残渣用硅胶柱色谱(庚烷/乙酸乙酯=4/1~2/1、然后氯仿/甲醇=20/1~10/1)纯化,由此以白色固体形态得到化合物(18b)(0.0826g、收率94%)。
[0186]1hnmr(400mhz,cdcl3):δ7.24
‑
7.17(m,4h),7.04(dt,1h),6.87
‑
6.81(m,4h),5.81(dt,1h),5.49(dd,1h),5.39(dd,1h),4.53
‑
4.23(m,4h),4.17
‑
4.12(m,1h),3.86
‑
3.72(m,8h),2.55
‑
2.46(m,1h),2.26
‑
2.19(m,2h),2.02
‑
1.93(m,2h),1.68
‑
1.03(m,17h),0.91
‑
0.81(m,6h)
[0187]
(步骤5
‑
7)
[0188]
(e)
‑7‑
((1r,2r,3r)
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑2‑
{(3s,5s,e)
‑3‑
[(4
‑
甲氧基苄基)氧基]
‑5‑
甲基壬
‑1‑
烯
‑1‑
基}
‑5‑
氧代环戊基)庚
‑2‑
烯酸(19b)
[0189]
将化合物(18b)(80.0mg、0.128mmol)溶解于二氯甲烷(1ml)中,在冰冷却下加入戴斯
‑
马丁试剂(dess
‑
martin periiodinane)(109mg、0.257mmol),在室温下搅拌2小时。向反应溶液中加入水、乙酸乙酯,用硅藻土进行过滤,减压馏去溶剂。将残渣用硅胶柱色谱(氯仿/甲醇=20/1~10/1、然后庚烷/乙酸乙酯=1/1)纯化,由此以无色透明油状物形态得到化合物(19b)(80.0mg、定量)。
[0190]1hnmr(400mhz,cdcl3):δ7.24
‑
7.18(m,4h),7.02(dt,1h),6.87
‑
6.80(m,4h),5.80
(dt,1h),5.61(dd,1h),5.52(dd,1h),4.56
‑
4.25(m,4h),3.92
‑
3.75(m,8h),2.77
‑
2.59(m,2h),2.28
‑
2.16(m,3h),2.02
‑
1.94(m,1h),1.69
‑
1.03(m,18h),0.90
‑
0.83(m,6h)
[0191]
(步骤5
‑
8)
[0192]
(e)
‑7‑
{(1r,2r,3r)
‑3‑
羟基
‑2‑
[(3s,5s,e)
‑3‑
羟基
‑5‑
甲基壬
‑1‑
烯
‑1‑
基]
‑5‑
氧代环戊基}庚
‑2‑
烯酸(9b)
[0193]
将化合物(19b)(10.0mg、16.0μmol)溶解于乙腈(0.5ml)与水(0.05ml)的混合溶剂中,在冰冷却下加入can(35.3mg、64.0μmol),在室温下搅拌30分钟。在冰冷却下加入乙酸乙酯,用饱和碳酸氢钠水溶液进行萃取。向水层中加入1mol/l盐酸,直至液体物性达到ph5,用乙酸乙酯进行萃取。将有机层用饱和食盐水洗涤,用无水硫酸镁干燥,减压馏去溶剂。将残渣用硅胶柱色谱(氯仿/甲醇=20/1~10/1)纯化,由此得到化合物(9b)(4.4mg、收率74%)。
[0194]1hnmr(400mhz,cdcl3):δ7.00(dt,1h),5.81(d,1h),5.66(dd,1h),5.56(dd,1h),4.25
‑
4.17(m,1h),4.11
‑
4.01(m,1h),3.40(br,1h),2.75(dd,1h),2.41
‑
2.32(m,1h),2.27
‑
2.14(m,3h),2.08
‑
1.97(m,1h),1.67
‑
1.11(m,16h),0.95
‑
0.84(m,6h)
[0195]
实施例10:拉坦前列素的制造方法
[0196][0197]
(步骤1)
[0198]
按照参考专利文献5中记载的方法,由市售的(
‑
)
‑
corey lactone
‑
benzoate(商品名)和2
‑
氧代
‑4‑
苯基丁基膦酸二甲酯制备化合物(3c)。与实施例1同样地操作,由化合物(3c)(20.0g、49mmol)得到化合物(4c)(非对映异构体过量率80%)。
[0199]
(步骤2)
[0200]
与实施例1同样地操作,由化合物(4c)得到化合物(7c)(20.0g、36mmol、收率74%(2步骤)、非对映异构体过量率99%)。
[0201]1hnmr(400mhz,cdcl3):δ8.22
‑
8.19(m,2h),8.07
‑
8.03(m,2h),7.91
‑
7.89(m,2h),7.52
‑
7.48(m,1h),7.37
‑
7.33(m,2h),7.26
‑
7.17(m,2h),7.16
‑
7.10(m,3h),5.74
‑
5.67(m,2h),5.51
‑
5.46(m,1h),5.25
‑
5.21(m,1h),5.05
‑
5.01(m,1h)2.88
‑
2.46(m,7h),2.23
‑
1.99(m,3h);
[0202]
13
cnmr(100mhz,cdcl3):δ176.2,165.9,163.8,150.6,140.8,135.6,133.4,132.5,130.9,130.7,129.6,129.4,128.7,128.5,128.4,126.3,123.6,83.1,78.6,77.5,77.1,76.8,75.3,54.0,42.5,37.5,35.8,34.8,31.6
[0203]
(步骤3)
[0204]
按照专利文献1中记载的方法,由化合物(7c)得到化合物(8c)。
[0205]
(步骤6)
[0206][0207]
按照专利文献5的实施例7中记载的方法,由化合物(8c)(5.0g,9.0mmol)得到化合物(10c)。进而按照专利文献4中记载的方法,由化合物(10c)得到化合物(9c)(1.1g、2.5mmol、收率28%(3个步骤))。
[0208]1hnmr(400mhz,cdcl3):δ7.29
‑
7.23(m,2h),7.21
‑
7.17(m,3h),5.48
‑
5.43(m,1h),5.41
‑
5.36(m,1h),4.99(m,1h),4.16(m,1h),3.94(m,1h),3.66(m,1h),2.82
‑
2.77(m,3h),2.70
‑
2.09(m,8h)1.90
‑
1.31(m,12h),1.22(d,6h);
[0209]
13
cnmr(100mhz,cdcl3):δ173.5,142.1,129.6,129.3,128.4,125.8,78.8,74.7,71.3,67.7,52.9,51.9,42.5,39.0,35.8,34.0,32.1,29.6,26.9,26.6,24.9,21.8。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。