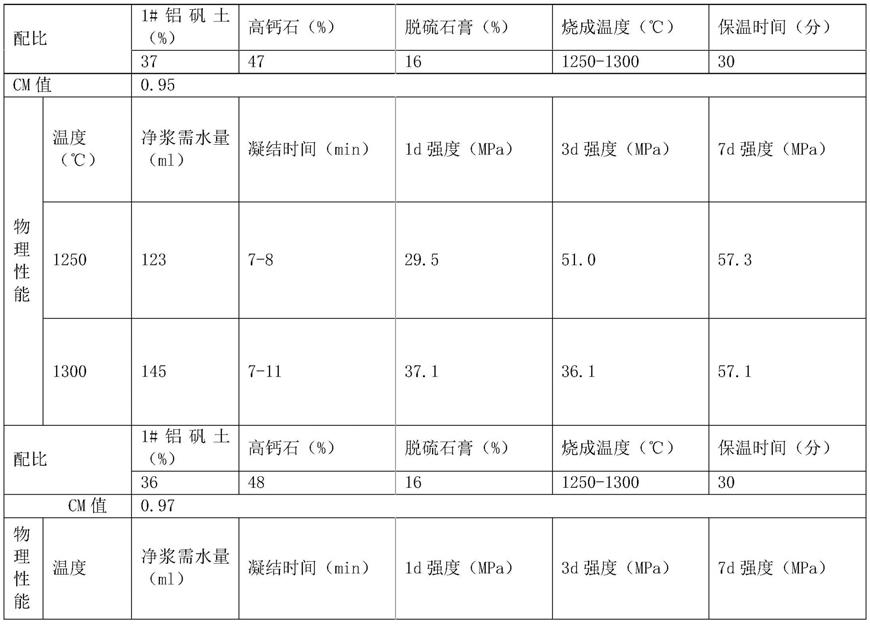
1.本发明涉及拟薄水铝石制备领域,具体涉及改性拟薄水铝石及其制备方法和改性氧化铝及加氢催化剂。
背景技术:
2.由于催化剂载体在催化反应进行过程中起到提供反应物以及产物扩散路径并为反应活性相的形成提供附着位的作用,因此载体表面与反应物及产物的吸附作用以及与活性组分的相互作用力对催化剂的性能会产生重要的影响。而这些相互作用力与氧化铝载体的比表面积以及表面的羟基数量和种类具有密切的关系。同时在对重质馏分油加氢处理过程中,由于原料中含有大量结构复杂、分子直径大、杂原子数量丰富的反应物分子,而且反应过程中由于金属沉积以及积炭作用的影响,使催化剂活性不断降低,因此要求催化剂不仅具有良好的反应活性,同时还需要具有优良的扩散性能以及容垢能力,为此,催化剂载体孔结构也会对催化剂性能起到重要影响。由此可见,具有高孔容、大比表面积而且表面羟基分布特殊的氧化铝载体在重油加氢催化剂制备过程中具有重要作用。
3.氧化铝、特别是γ-氧化铝,因其具有较好孔结构、比表面和耐热稳定性,常作为载体用于催化剂的制备。氧化铝的前身物为水合氧化铝,如拟薄水铝石,其粒子大小、形貌、结晶度和杂晶含量等都对氧化铝载体的孔容、孔分布、比表面积等性质产生影响。现有技术中,通过对水合氧化铝的粒子大小、形貌、结晶度等性质的调变,可以获取可满足特定需要的氧化铝载体。
4.作为氧化铝载体原料的拟薄水铝石一般由如下方法进行制备:(1)碱沉淀法,即酸化铝盐与碱中和。用碱从酸化铝盐溶液沉淀出一水合氧化铝,再通过老化、洗涤、干燥等过程得到拟薄水铝石产品,该法常被称为碱沉淀(酸法),如氨水中和三氯化铝的方法;(2)酸沉淀法,即强酸或强酸的铝盐中和铝酸盐。先用酸从铝酸盐溶液中沉淀出一水合氧化铝,再通过老化、洗涤、干燥等过程得到拟薄水铝石产品,常被称为酸沉淀(碱法),目前最为常用方法包括:co2气体中和偏铝酸钠的方法、硫酸铝中和偏铝酸钠的方法;(3)烷氧基铝水解法,将烷氧基铝与水发生水解反应生成一水合氧化铝,再经老化、过滤、干燥得到拟薄水铝石产品。在拟薄水铝石的制备过程一般都由晶粒生成(中和沉淀或者水解过程)、晶粒生长(老化过程)、洗涤、干燥等过程组成。因此,晶粒生成、晶粒生长的工艺条件会对晶粒生成的数量和生长速度产生影响,各种拟薄水铝石的制备工艺都提出了各自的工艺条件,控制产品的晶粒大小、结晶度,以达到控制产品孔容和比表面积等物理性质的目的。
5.在氧化铝中引入磷,可以改变载体的孔结构、表面酸性和热稳定性,从而可以提高加氢催化剂的活性。
6.一种方法是先由拟薄水铝石粉体,通过成型、焙烧制备氧化铝载体,再通过浸渍法在氧化铝载体上引入磷,制备磷改性氧化铝。采用浸渍法制备磷改性活性氧化铝,可以提高氧化铝的热稳定性,但是用磷酸浸渍氧化铝,部分氧化铝溶解在磷酸溶液中,与磷酸根发生反应生成磷酸铝,沉积在氧化铝孔道中,会堵塞孔道,造成比表面积和孔容的下降。
7.一种方法是在拟薄水铝石成型过程中添加含磷化合物,随后焙烧成型化合物制备磷改性氧化铝。cn103721732a公开了一种加磷改性拟薄水铝石催化剂载体材料及制备方法。在中和反应釜1中,加入氧化铝浓度45-55g/l的硫酸铝溶液和氧化铝浓度为200-250g/l、苛性比为1.1-1.3的偏铝酸钠溶液,控制ph为6.0-8.0,温度为50-70℃;中和反应釜1的浆液通过溢流反应管流入中和反应釜2,同时向中和反应釜2中加入浓度为100-200g/l的碳酸钠溶液,控制ph为8.5-10.0,控制反应温度在50-70℃;中和反应釜2的浆液通过溢流反应管流入老化反应釜,老化反应釜内浆液温度80-95℃,老化2小时;根据中和反应釜1反应过程中投入氧化铝的质量,计算向老化反应釜加入五氧化二磷浓度为50-150g/l的磷酸溶液的体积,加入磷酸的五氧化二磷含量为氧化铝含量的3%-5%;老化完成后洗涤、干燥,得到含磷拟薄水铝石。
8.尽管上述文献公开了多种不同的制备含磷拟薄水铝石的方法,并且获得的拟薄水铝石的性能在某些方面较优异,然而,由它们制得的氧化铝用作催化剂载体时,该催化剂的性能有待进一步提高。
技术实现要素:
9.本发明的目的是为了克服由现有技术的拟薄水铝石制得的氧化铝用作催化剂载体时,该催化剂的加氢活性有待进一步提高的缺陷,提供一种改性拟薄水铝石及其制备方法和改性氧化铝及加氢催化剂。采用本发明提供的改性拟薄水铝石制得的载体得到的催化剂具有较好的加氢活性。
10.本发明的发明人在研究过程中发现,在拟薄水铝石的制备过程中,通过在原料中添加含磷化合物和非金属助剂元素,在沉淀反应或者水解反应过程中添加晶粒生长调节剂,且控制沉淀反应或者水解反应的ph为4-7,随后再调节ph至7-10.5老化的方法,加强晶粒生长方式的调节,从而可以制备出1.7≤h≤4的改性拟薄水铝石产品,优选1.9≤h≤4,更优选2.2≤h≤3.5,可以有效提高以该改性拟薄水铝石焙烧后得到的改性氧化铝作为载体的催化剂的加氢活性。而上述现有技术制备的含磷拟薄水铝石由于未对h进行控制,h一般为0.85-1.65。本发明的改性拟薄水铝石由于具有1.7≤h≤4的特征,优选1.9≤h≤4,更优选2.2≤h≤3.5,因此用作加氢催化剂的载体的前躯体时,能提高催化剂的加氢性能。
11.为了实现上述目的,本发明第一方面提供一种改性拟薄水铝石,该改性拟薄水铝石中含有磷元素和非金属助剂元素,该改性拟薄水铝石的h满足1.7≤h≤4,其中h=d(031)/d(020),所述d(031)表示拟薄水铝石晶粒的xrd谱图中031峰所代表的晶面的晶粒尺寸,d(020)表示拟薄水铝石晶粒的xrd谱图中020峰所代表的晶面的晶粒尺寸,所述031峰是指xrd谱图中2θ为34-43
°
的峰,所述020峰是指xrd谱图中2θ为10-15
°
的峰,d=kλ/(bcosθ),k为scherrer常数,λ为靶型材料的衍射波长,b为衍射峰的半峰宽,2θ为衍射峰的位置。
12.优选地,该改性拟薄水铝石的h满足1.9≤h≤4,优选满足2.2≤h≤3.5。
13.本发明第二方面提供一种改性拟薄水铝石的制备方法,该方法包括以下步骤:
14.(1)将无机含铝化合物溶液与酸或碱接触进行沉淀反应,或者将有机含铝化合物与水接触进行水解反应,得到改性水合氧化铝;
15.(2)将上述得到的改性水合氧化铝在ph为7-10.5条件下进行老化;
16.步骤(1)所述沉淀反应或者所述水解反应在晶粒生长调节剂以及含磷化合物和含
非金属助剂化合物存在下、ph为4-7条件下进行;所述晶粒生长调节剂为能够调节晶粒在不同晶面上的生长速度的物质。
17.本发明第三方面提供一种改性氧化铝,该改性氧化铝由改性拟薄水铝石焙烧得到,所述改性拟薄水铝石为前述第一方面的改性拟薄水铝石或者前述第二方面所述的方法制得的改性拟薄水铝石。
18.本发明第四方面提供一种改性氧化铝,该改性氧化铝中含有磷元素和非金属助剂元素,该改性氧化铝的ir谱图中,(i
3670
i
3580
)/(i
3770
i
3720
)为1.9-3.5,其中,i
3670
为3670cm-1
处峰高,i
3580
为3580cm-1
处峰高,i
3770
为3770cm-1
处峰高,i
3720
为3720cm-1
处峰高。
19.本发明第五方面提供一种加氢催化剂,该催化剂包括载体和负载在载体上的活性金属组分,所述载体为前述第三方面或者第四方面所述的改性氧化铝。
20.与现有技术相比,本发明提供的改性拟薄水铝石由于具有1.7≤h≤4的特征,因而该改性拟薄水铝石经焙烧后的改性氧化铝更加适合用作重油加氢催化剂载体,所得催化剂具有更优异的加氢活性和高稳定性。本发明提供的改性拟薄水铝石的制备方法通过添加含磷化合物、含非金属助剂化合物、晶粒生长调节剂、制备过程ph的分段控制,使得到的改性拟薄水铝石具有1.7≤h≤4的特征。该改性拟薄水铝石经焙烧后的改性氧化铝具有特定的表面羟基分布,该改性氧化铝的ir谱图中,(i
3670
i
3580
)/(i
3770
i
3720
)为1.9-3.5;其中,i
3670
为3670cm-1
处峰高,i
3580
为3580cm-1
处峰高,i
3770
为3770cm-1
处峰高,i
3720
为3720cm-1
处峰高,更加适合用作催化剂载体,所得催化剂具有更优异的重油加氢活性和高稳定性。
具体实施方式
21.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
22.本发明第一方面提供一种改性拟薄水铝石,该改性拟薄水铝石中含有磷元素和非金属助剂元素,该改性拟薄水铝石的h满足1.7≤h≤4,其中h=d(031)/d(020),所述d(031)表示拟薄水铝石晶粒的xrd谱图中031峰所代表的晶面的晶粒尺寸,d(020)表示拟薄水铝石晶粒的xrd谱图中020峰所代表的晶面的晶粒尺寸,所述031峰是指xrd谱图中2θ为34-43
°
的峰,所述020峰是指xrd谱图中2θ为10-15
°
的峰,d=kλ/(bcosθ),k为scherrer常数,λ为靶型材料的衍射波长,b为衍射峰的半峰宽,2θ为衍射峰的位置。
23.在本发明中,对于不同的衍射峰,b和2θ均取与之相应的峰的值,例如,计算d(031)时,d(031)=kλ/(bcosθ),其中b为031衍射峰的半峰宽,2θ为031衍射峰的位置;计算d(020)时,d(020)=kλ/(bcosθ),其中b为020衍射峰的半峰宽,2θ为020衍射峰的位置。本发明中,所述非金属助剂元素不包括磷元素。
24.优选情况下,该改性拟薄水铝石的h满足1.9≤h≤4,更优选满足2.2≤h≤3.5。在该优选范围内,则所得催化剂的加氢活性更优。
25.h满足上述规定的改性拟薄水铝石通过焙烧制得的改性氧化铝具有特定的羟基分布,更有利于提高催化剂的脱硫性能。而现有技术制得的拟薄水铝石中,h一般为0.85-1.65。
26.本发明提供的改性拟薄水铝石的相对结晶度(以condea公司的商业sb粉为基准)一般在45-77%范围内,优选为65-77%范围内。
27.优选地,所述非金属助剂元素包括氟元素和/或硅元素。
28.本发明提供的改性拟薄水铝石中含有磷元素和非金属助剂元素,优选地,以改性拟薄水铝石的干基总量为基准,al2o3的含量为79-98.9重量%,优选为85-97.5重量%;p2o5的含量为1-6重量%,优选为2-5重量%,所述非金属助剂元素含量为0.1-15重量%,优选为0.5-10重量%。
29.本发明中,当所述非金属助剂元素为f元素时,所述非金属助剂元素含量以f元素计;当所述非金属助剂元素为si元素时,所述非金属助剂元素含量以sio2计。
30.在本发明中,改性拟薄水铝石的晶体结构采用德国西门子公司的d5005型x射线衍射仪测定,cukα辐射,44千伏,40毫安,扫描速度为2
°
/分钟。
31.本发明提供的改性拟薄水铝石含有磷元素和非金属助剂元素,且具有特定的晶体结构,含有本发明提供的改性拟薄水铝石制得载体的催化剂表现出优异的加氢活性和高稳定性。
32.本发明第二方面提供一种改性拟薄水铝石的制备方法,该方法包括以下步骤:
33.(1)将无机含铝化合物溶液与酸或碱接触进行沉淀反应,或者将有机含铝化合物与水接触进行水解反应,得到改性水合氧化铝;
34.(2)将上述得到的改性水合氧化铝在ph为7-10.5条件下进行老化;
35.步骤(1)所述沉淀反应或者所述水解反应在晶粒生长调节剂以及含磷化合物和含非金属助剂化合物存在下、ph为4-7条件下进行;所述晶粒生长调节剂为能够调节晶粒在不同晶面上的生长速度的物质。
36.本发明提供的方法中,所述沉淀反应或者所述水解反应在晶粒生长调节剂以及含磷化合物和含非金属助剂化合物存在下、ph为4-7条件下进行,既可以满足改性水合氧化铝的沉淀,又保持较低的ph条件,以避免高ph下改性拟薄水铝石晶粒生长过快,加强磷及生长调节剂对改性拟薄水铝石生长的共同调节作用。改性水合氧化铝的生成、老化整个过程改性拟薄水铝石的晶粒生长,都在含磷化合物、含非金属助剂化合物和晶粒调节剂共同存在下进行,使得制得的改性拟薄水铝石具有特殊的晶体结构,特别适用于作用重油加氢催化剂的载体前驱体。
37.根据本发明的一种具体实施方式,步骤(1)包括:将无机含铝化合物溶液、含磷化合物、含非金属助剂化合物、晶粒生长调节剂以及酸或碱接触进行沉淀反应,或者,将有机含铝化合物、含磷化合物、含非金属助剂化合物、晶粒生长调节剂与水进行水解反应;控制所述沉淀反应或者所述水解反应的ph为4-7。
38.根据本发明的一种优选实施方式,步骤(1)所述沉淀反应或者所述水解反应在晶粒生长调节剂以及含磷化合物和含非金属助剂化合物存在下、ph为4-6.5条件下进行。使得沉淀反应或者水解反应在上述优选ph下进行,更有利于提高制得的载体在重油加氢中的活性。
39.对所述沉淀反应和水解反应的除ph外其它条件没有特别限定。本发明中,优选地,所述沉淀反应和所述水解反应的温度各自独立地为30-90℃。
40.本发明中,对所述沉淀反应的条件选择范围较宽,优选地,所述沉淀反应的条件包
括:反应的温度为40-90℃,反应的时间为10-60分钟。进一步优选地,所述沉淀反应的条件包括:反应的温度为45-80℃,反应的时间为10-30分钟。
41.本发明对所述水解反应的条件没有特别限定,只要使得水与有机含铝化合物接触发生水解反应生成改性水合氧化铝即可。本发明对所述水解反应过程中水的用量选择范围较宽,只要使得水与有机含铝化合物的摩尔比为大于化学计量比即可。具体发生水解的条件为本领域技术人员所公知。优选地,所述水解反应的条件包括:反应的温度为40-90℃,优选为45-80℃,反应的时间为2-30小时,优选为2-20小时。
42.在本发明中,所述晶粒生长调节剂为能够调节晶粒在不同晶面上的生长速度的物质,优选为能够调节晶粒在020晶面和031晶面的生长速度的物质。例如,可以为各种能与水合氧化铝发生强吸附作用的物质,优选地,所述晶粒生长调节剂为多羟糖醇及其羧酸盐和硫酸盐中的至少一种;进一步优选地,所述晶粒生长调节剂选自山梨糖醇、葡萄糖、葡萄糖酸、葡萄糖酸盐、核糖醇、核糖酸、核糖酸盐和硫酸盐中的至少一种。所述葡萄糖酸盐、核糖酸盐和硫酸盐各自可以为它们的可溶性盐,例如,可以为钾盐、钠盐和锂盐中的一种或几种。
43.本发明中,对所述晶粒生长调节剂的加入方式没有特别限定,可以将晶粒生长调节剂单独加入,也可以预先将晶粒生长调节剂与其中的一种或几种原料混合,然后再将含有晶粒生长调节剂的原料进行反应。
44.本发明对晶粒生长调节剂的用量没有特别的限定,优选地,以氧化铝计,所述沉淀反应中晶粒生长调节剂的用量为无机含铝反应物重量的1-10重量%,进一步优选为1.5-8.5重量%,更进一步优选2-6重量%。
45.优选地,以氧化铝计,所述水解反应中,所述晶粒生长调节剂的用量为有机含铝化合物重量的1-10重量%,优选为1.5-8.5重量%,更进一步优选为2-6重量%。
46.除非特别说明,本发明中,所述晶粒生长调节剂的用量均分别以有机含铝化合物、无机含铝化合物中对应的氧化铝的重量为基准进行计算。
47.在本发明中,对所述含磷化合物、含非金属助剂化合物的加入方式没有特别限定,可以将含磷化合物(或者配制成含磷化合物水溶液)、含非金属助剂化合物(或者配制成含非金属助剂化合物水溶液)分别单独加入,也可以预先将含磷化合物(或者其水溶液)、含非金属助剂化合物(或者配制成其水溶液)分别与其中的一种或几种原料混合,然后再将含有含磷化合物、含非金属助剂化合物的原料进行反应,只要保证所述沉淀反应或者水解反应在含磷化合物和含非金属助剂化合物存在下进行即可。本发明提供的制备方法可以保证含磷化合物和含非金属助剂化合物对晶粒生长的调节效果。
48.本发明对所述含磷化合物的种类选择范围较宽,可以为水溶性无机含磷化合物,优选情况下,所述含磷化合物选自磷酸、磷酸铵、磷酸氢铵、磷酸氢二铵、磷酸钠和磷酸钾中的至少一种。
49.为了更好发挥含磷化合物和含非金属助剂化合物对晶粒生长的调节效果,优选地,所述含磷化合物和含非金属助剂化合物的用量使得制得的改性拟薄水铝石中,以改性拟薄水铝石的干基总量为基准,p2o5的含量为1-6重量%,优选为2-5重量%,所述非金属助剂元素含量为0.1-15重量%,优选为0.5-10重量%。
50.本发明对所述含非金属助剂化合物没有特别限定,可以为含本领域公知的非金属
助剂的化合物;优选地,所述非金属助剂包括含氟化合物和/或含硅化合物。
51.优选地,所述含氟化合物为氟化铵、氟化氢铵、氢氟酸、氟化钠和氟化钙中的至少一种。
52.优选地,所述含硅化合物选自氧化硅、硅溶胶、水玻璃和硅酸钠中的至少一种。
53.需要说明的是,本发明在研究过程中发现,晶粒生长调节剂以及含磷化合物和含非金属助剂化合物在所述沉淀反应或者所述水解反应过程中加入,更有利于对晶粒在020晶面和031晶面的生长速度进行调节,以使得h满足1.7≤h≤4,优选满足1.9≤h≤4,更优选满足2.2≤h≤3.5。在所述沉淀反应或者所述水解反应过程中加入晶粒生长调节剂以及含磷化合物和含非金属助剂化合物,使得在后进行的老化反应同样在晶粒生长调节剂以及含磷化合物和含非金属助剂化合物存在下进行。优选情况下,所述老化过程中不额外加入晶粒生长调节剂以及含磷化合物和含非金属助剂化合物。
54.按照本发明提供的方法,所述无机含铝化合物优选为铝盐和/或铝酸盐。相应的,所述无机含铝化合物溶液可以是各种铝盐溶液和/或铝酸盐溶液,所述铝盐溶液可以是各种铝盐溶液,例如可以是硫酸铝、氯化铝、硝酸铝中的一种或几种的水溶液。因为价格低,优选硫酸铝溶液和/或氯化铝溶液。铝盐可以单独使用也可以两种或者多种混合后使用。所述铝酸盐溶液是任意的铝酸盐溶液,如铝酸钠溶液和/或铝酸钾溶液。因为其获得容易而且价格低,优选为铝酸钠溶液。铝酸盐溶液也可以单独或者混合使用。
55.对所述无机含铝化合物溶液的浓度没有特别限定,优选地,以氧化铝计,所述无机含铝化合物溶液的浓度为20-200克/升。
56.所述酸可以是各种质子酸或在水介质中呈酸性的氧化物,例如,可以是硫酸、盐酸、硝酸、碳酸、磷酸、甲酸、乙酸、柠檬酸和草酸中的至少一种,优选地,所述质子酸选自硝酸、硫酸和盐酸中的至少一种。所述碳酸可以通过向铝盐溶液和/或铝酸盐溶液中通入二氧化碳而原位产生。所述酸可以以溶液的形式引入,对所述酸溶液的浓度没有特别限定,优选h
的浓度为0.2-2摩尔/升。
57.所述的碱可以为氢氧化物或在水介质中水解使水溶液呈碱性的盐,优选地,所述氢氧化物选自氨水、氢氧化钠和氢氧化钾中的至少一种;优选地,所述盐选自偏铝酸钠、偏铝酸钾、碳酸氢铵、碳酸铵、碳酸氢钠、碳酸钠、碳酸氢钾和碳酸钾中的至少一种。所述碱可以以溶液的形式引入,对所述碱溶液的浓度没有特别限定,优选oh-的浓度为0.2-4摩尔/升。当以偏铝酸钠和/或偏铝酸钾作为碱时,计算所述晶粒生长调节剂和含磷化合物的用量时,也考虑偏铝酸钠和/或偏铝酸钾中相应的氧化铝的量。
58.按照本发明提供的方法,所述有机含铝化合物可以是各种能与水发生水解反应,产生水合氧化铝沉淀的烷氧基铝中的至少一种,例如可以是异丙醇铝、异丁醇铝、三异丙氧基铝、三特丁氧基铝和异辛醇铝中的至少一种。
59.具体地,为了调控水解反应的ph,可以在水解反应中引入酸或碱,所述酸或碱的引入方式和种类可以如上文所述,在此不再赘述。
60.其中,通过对反应物中碱或酸的用量控制ph而使铝沉淀的方法为本领域技术人员所公知,在此不再赘述。
61.本发明对步骤(2)所述老化的条件选择范围较宽,只要保证在ph为7-10.5条件下进行即可。由于步骤(1)所述沉淀反应或者所述水解反应在ph为4-7条件下进行,优选在进
行老化之前,引入碱以调节老化反应的ph。所述碱的引入方式和种类可以如上文所述。
62.优选地,步骤(2)所述老化在ph为8-10条件下进行。
63.本发明对步骤(2)所述老化除了ph外的条件选择范围较宽,优选地,所述老化的温度为50-95℃,优选为55-90℃。所述老化的时间根据老化温度进行适当选择,优选地,老化时间为0.5-8小时,优选为2-6小时。
64.本发明还包括在老化反应后,对老化产物进行分离、洗涤和干燥。按照本发明提供的方法,所述分离可以为本领域的公知技术,如过滤或离心分离的方法。所述洗涤和干燥的方法可以为制备拟薄水铝石中常用的方法,例如,所述洗涤用剂可以为水,所述干燥可以为烘干、鼓风干燥、喷雾干燥和闪蒸干燥中的至少一种。干燥温度可以为100-350℃,优选为120-300℃。
65.根据本发明的一种优选实施方式,所述制备方法包括如下步骤:
66.(1)将含磷化合物、含非金属助剂化合物和晶粒生长调节剂的无机含铝化合物溶液与碱溶液或酸溶液并流或者间歇式加入到反应容器中进行沉淀反应,得到改性水合氧化铝浆液;或者,在去离子水中加入含磷化合物、含非金属助剂化合物和晶粒生长调节剂与烷氧基铝进行水解反应,得到改性水合氧化铝浆液,并通过酸溶液或者碱溶液的用量,使得沉淀反应或者水解反应在ph为4-7,优选4-6.5条件下进行;
67.(2)将步骤(1)得到的改性水合氧化铝浆液,加入碱性溶液调节ph为7-10.5后,于50-95℃老化0.5-8小时;
68.(3)过滤、洗涤步骤(2)得到的产物;
69.(4)干燥步骤(3)得到的产物,得到本发明提供的改性拟薄水铝石。
70.本发明第三方面提供一种改性氧化铝,该改性氧化铝由改性拟薄水铝石焙烧得到,所述改性拟薄水铝石为前述第一方面的改性拟薄水铝石或者前述第二方面所述的方法制得的改性拟薄水铝石。
71.在本发明的一种优选实施方式中,所述改性氧化铝由改性拟薄水铝石依次进行任选的成型、干燥以及焙烧得到。
72.本发明对所述成型条件、干燥条件和焙烧条件没有特别限定,可以为本领域惯常的方式。所述成型方法可以为滚球、压片和挤条成型中的至少一种,优选挤条成型,之后进行干燥并焙烧;所述成型后的形状可以为三叶草形、蝶形、圆柱形、中空圆柱形、四叶形、五叶形、球形等。为保证所述成型顺利进行,还可以加入水、助挤剂和/或胶粘剂,并可选地加入扩孔剂,所述助挤剂、胶溶剂和扩孔剂的种类和用量为本领域技术人员公知,例如,常见的助挤剂可以选自田菁粉、甲基纤维素、淀粉、聚乙烯醇和聚乙醇中的至少一种,所述胶溶剂可以为有机酸和/或有机酸,所述扩孔剂可以为淀粉、合成纤维素、聚合醇和表面活性剂中的至少一种。其中,所述合成纤维素优选为羟甲基纤维素、甲基纤维素、乙基纤维素和羟基纤维脂肪醇聚乙烯醚中的至少一种;所述聚合醇优选为聚乙二醇、聚丙醇和聚乙烯醇中的至少一种;所述表面活性剂优选为脂肪醇聚乙烯醚、脂肪醇酰胺及其衍生物、分子量为200-10000的丙烯醇共聚物和顺丁烯酸共聚物中的至少一种。所述干燥的条件优选包括:干燥温度40-350℃,更优选为100-200℃;干燥时间1-24小时,更优选为2-12小时。
73.本发明对所述焙烧的条件没有特别的限定,优选地,所述焙烧的条件包括:温度为350-1000℃,优选为400-800℃,时间为1-10小时,优选为2-6小时。
74.本发明第四方面提供一种改性氧化铝,该改性氧化铝中含有磷元素和非金属助剂元素,该改性氧化铝的ir谱图中,(i
3670
i
3580
)/(i
3770
i
3720
)为1.9-3.5,优选为2-3.3;其中,i
3670
为3670cm-1
处峰高,i
3580
为3580cm-1
处峰高,i
3770
为3770cm-1
处峰高,i
3720
为3720cm-1
处峰高。该非金属助剂元素与本发明前述第一方面提供的非金属助剂元素相同,在此不再赘述。
75.根据本发明第四方面提供的改性氧化铝,该改性氧化铝由改性拟薄水铝石焙烧得到,其中,所述改性拟薄水铝石为前述第一方面所述的改性拟薄水铝石或者前述第二方面所述的方法制得的改性拟薄水铝石。
76.本发明提供的改性氧化铝具有特定的表面羟基分布,将其作为载体用于重油加氢催化剂,使得催化剂具有较高的加氢活性和高稳定性。
77.所述ir谱图通过美国nicolet公司nicolet 870型傅里叶红外光谱仪测定得到。具体包括:将样品压成自支撑片,置于红外池中,在真空条件下450℃处理样品3h,测定样品的红外光谱。根据谱图上3670cm-1
处峰高、3580cm-1
处峰高、3770cm-1
处峰高、3720cm-1
处峰高的值计算(i
3670
i
3580
)/(i
3770
i
3720
)的值。现有技术的氧化铝载体(i
3670
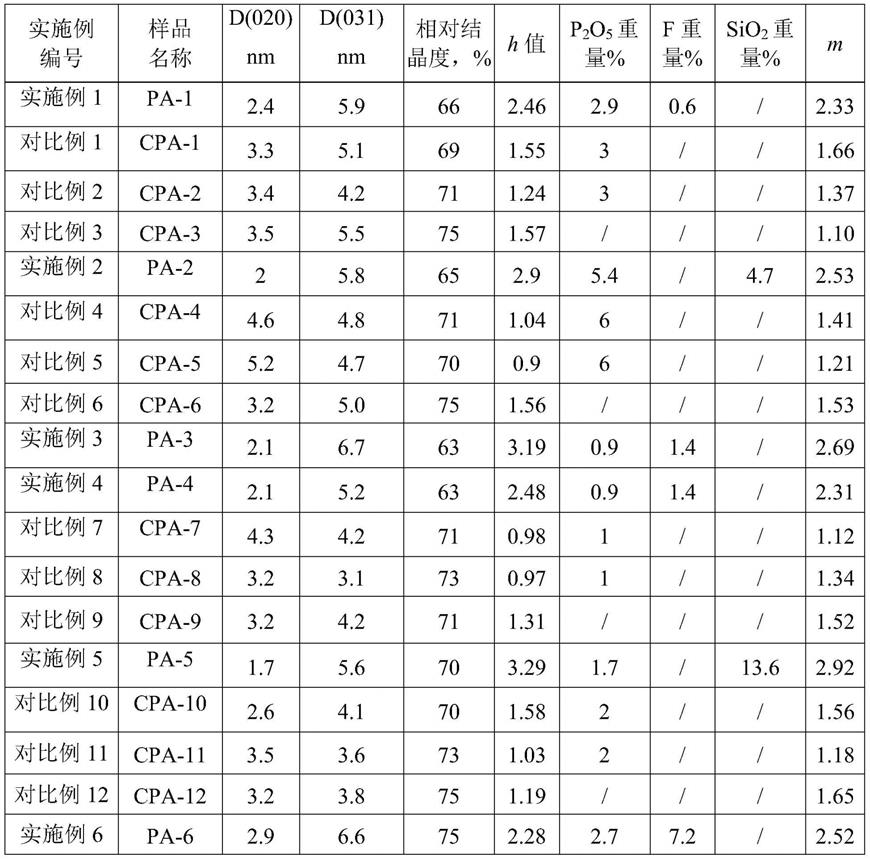
i
3580
)/(i
3770
i
3720
)的值一般低于1.8。
78.根据本发明,优选地,所述改性氧化铝的氮吸附法孔容为0.7-1.6毫升/克,bet氮吸附法比表面积为250-380平方米/克,可几孔直径为8-16纳米。所述可几孔直径指孔分布曲线中,曲线的最高点所对应的直径大小。本发明提供的改性氧化铝孔容和比表面积均较大。
79.本发明提供的改性氧化铝可以作为各种吸附剂、催化剂载体和催化剂的基质使用。
80.本发明第五方面提供一种加氢催化剂,该催化剂包括载体和负载在载体上的活性金属组分,所述载体为前述第三方面或第四方面所述的改性氧化铝。
81.根据本发明,对所述活性金属组分的种类及其含量没有特别限制,可以为本领域常用于烃油加氢处理催化剂的活性金属组分与含量;优选地,所述活性金属组分选自vib族金属组分和/或viii族金属组分。本发明对所述vib族金属组分和viii族金属组分没有特别限制,所述vib族金属组分优选为mo和/或w,所述viii族金属组分优选为co和/或ni。
82.优选地,以所述加氢催化剂的总量为基准,所述载体的含量为40-94重量%,以氧化物计,所述vib族金属组分的含量为5-45重量%,所述viii族金属组分的含量为1-15重量%。
83.根据本发明,进一步地,所述vib族金属化合物和viii族金属化合物各自独立地选自它们的可溶性化合物中的至少一种(包括在助溶剂存在下可溶于水的相应金属化合物)。具体地,所述vib族金属化合物,以钼为例,可以选自含钼金属的盐和/或氧化物,例如可以选自氧化钼、钼酸盐、仲钼酸盐、磷钼酸盐中的至少一种,优选氧化钼、钼酸铵、仲钼酸铵、磷钼酸中的至少一种;viii族金属化合物,以钴为例,可以选自硝酸钴、醋酸钴、碱式碳酸钴、氯化钴中的至少一种,优选硝酸钴和/或碱式碳酸钴,以镍为例,可以选自含镍的盐、氧化物和氢氧化物中的至少一种,例如可以选自镍的硝酸盐、氯化物、甲酸盐、乙酸盐、磷酸盐、柠檬酸盐、草酸盐、碳酸盐、碱式碳酸盐、氢氧化物、磷化物、硫化物、铝酸盐、钼酸盐和氧化物中的至少一种,优选镍的草酸盐、碳酸盐、碱式碳酸盐、氢氧化物、磷酸盐和氧化物中的至少一种,更优选硝酸镍、醋酸镍、碱式碳酸镍、氯化镍和碳酸镍中的至少一种。
84.根据本发明,在加氢催化剂制备过程中,如在所述vib族金属化合物和viii族金属化合物的可溶性化合物制备过程中,本发明还可以含有有机添加物。本发明对所述有机添加物的引入方式没有特别限制,所述有机添加物可以以任意方式引入,例如可以和所述viii族金属一起引入,也可以和所述vib族金属元素一起引入,还可以在引入所述viii族和/或vib族金属元素之后引入,也可以在引入所述viii族和/或vib族元素之前引入。本发明对所述有机添加物的种类没有特别限制,所述有机添加物选自含氧和/或含氮的有机物中的至少一种,所述含氧有机物选自有机醇和/或有机酸,所述含氮有机物选自有机胺和有机胺盐中的至少一种;具体地,所述含氧有机物选自乙二醇、丙三醇、聚乙二醇(分子量为200-1500)、二乙二醇、丁二醇、乙酸、马来酸、草酸、氨基三乙酸、1,2-环己烷二胺四乙酸、柠檬酸、酒石酸、苹果酸中的至少一种,优选乙二醇、丙三醇、聚乙二醇和柠檬酸中的至少一种;所述含氮有机物选自乙二胺、二亚乙基三胺、环己二胺四乙酸、氨基乙酸、次氮基三乙酸、edta及其胺盐中的至少一种,优选edta和/或次氮基三乙酸。
85.本发明提供的加氢催化剂中还可以含有任何不影响本发明加氢催化剂的性能或能改善本发明加氢催化剂性能的助剂,如可以含有iia、iiia、iva、va、viia、iib、iiib族元素和稀土金属元素中的至少一种,优选为硼、氟、硅、锌、钙、钛、镧、铈中的至少一种,以催化剂为基准,以单质元素计的所述助剂的含量不超过10重量%,优选为0.5-6重量%。
86.本发明对所述加氢催化剂的制备方法没有特别的限定,只要将所述加氢活性金属组分负载在复合催化剂上即可,可以是本领域中任意的惯常方法,例如可以为混捏法、干混法、浸渍法;优选地,将所述加氢活性金属组分负载在含磷氧化铝上的方法包括采用含有至少一种vib族金属化合物和至少一种viii族金属化合物的浸渍液浸渍所述含磷氧化铝,然后进行干燥和焙烧。进一步地,本发明对所述浸渍的方法和浸渍时间不作特别限定,所述浸渍的方法,根据浸渍液用量不同可以为过量液浸渍、孔饱和法浸渍和多次浸渍法等,根据浸渍实现的方式不同可以为浸泡法、喷淋浸渍等;所述浸渍时间优选为0.5-3小时。更进一步地,通过对所述浸渍液的浓度、用量或载体用量的调节和控制,可以制备出特定含量的加氢催化剂,这是本领域技术人员所公知的。
87.根据本发明,对所述加氢活性金属组分负载在催化剂上的方法中的干燥条件和焙烧条件不作特别限定,优选地,干燥的条件包括:干燥温度为80-200℃,优选为100-150℃;干燥时间为1-8小时,优选为2-6小时。本发明对所述干燥的方式不作特别限定,所述干燥可以为烘干、鼓风干燥、喷雾干燥和闪蒸干燥中的至少一种。优选地,焙烧的条件包括:焙烧温度为200-700℃,优选为350-600℃;焙烧时间为1-10小时,优选为2-8小时。根据本发明,对所述焙烧和所述干燥的气氛没有特别限制,可以是空气、氧气和氮气的至少一种,优选为空气。
88.根据本发明的一种优选实施方式,所述加氢催化剂的制备方法包括:将含有活性金属组分的浸渍液浸渍所述改性氧化铝,然后于80-200℃下干燥1-8小时,再于200-700℃下焙烧1-10小时。
89.本发明提供的加氢催化剂可以单独使用,也可以与其他催化剂组合使用。
90.根据本发明,加氢催化剂在使用之前,可以按照本领域中的常规方法进行预硫化,以使其上负载的活性金属组分转化为金属硫化物组分;预硫化方法可以为:将加氢催化剂在氢气存在下,于140-400℃条件下用硫、硫化氢或含硫原料进行预硫化。这种预硫化可在
器外进行,也可在器内原位硫化。
91.本发明对加氢催化剂应用时的加氢条件没有特别的限定,可以采用本领域中通常的反应条件;优选地,反应温度为200-420℃,进一步优选为220-400℃,压力为2-18mpa,进一步优选为2-16mpa,液时体积空速0.1-10小时-1
、进一步优选为0.15-6小时-1
,氢油体积比为50-5000、进一步优选为50-4000。
92.本发明对加氢催化剂应用时的加氢处理反应装置没有特别限制,可以为任何足以使原料油在加氢处理反应条件下与加氢催化剂进行接触反应的反应器,例如固定床反应器、浆态床反应器、移动床反应器或沸腾床反应器。
93.本发明对加氢催化剂的应用对象没有特别限制,可以直接用于加工各类烃油原料,以对其进行加氢改质或加氢裂化。所述烃油原料可以是各种重质矿物油或合成油或它们的混合馏分油,例如可以为选自原油、馏分油、溶剂精制油、蜡膏、蜡下油、费托合成油、煤液化油、轻脱沥青油和重脱沥青油中的至少一种;特别适合用于汽油、柴油、蜡油、润滑油、煤油、石脑油、常压渣油、减压渣油、石油蜡和费托合成油中至少一种的加氢处理。
94.以下将通过实施例对本发明进行详细描述。以下实施例中,xrd在simens d5005型x光衍射仪上测定,cukα辐射,44千伏,40毫安,扫描速度为2
°
/分钟。根据scherrer公式:d=kλ/(bcosθ)(d为晶粒尺寸,λ为靶型材料的衍射波长,b为校正过的衍射峰的半峰宽,2θ为衍射峰的位置)分别以2θ为10-15
°
峰的参数计算出(020)的晶粒大小为d(020)、以2θ为34-43
°
峰的参数计算出(031)的晶粒大小为d(031),并计算出h=d(031)/d(020)。
95.所述ir谱图通过美国nicolet公司nicolet 870型傅里叶红外光谱仪测定得到。具体包括:将样品压成自支撑片,置于红外池中,在真空条件下450℃处理样品3h,测定样品的红外光谱。根据谱图上3670cm-1
处峰高、3580cm-1
处峰高、3770cm-1
处峰高、3720cm-1
处峰高的值计算(i
3670
i
3580
)/(i
3770
i
3720
)的值。
96.实施例1
97.该实施例用于说明本发明提供的改性拟薄水铝石、改性氧化铝(即载体)和加氢催化剂及其制备方法。
98.(1)制备水合氧化铝pa1:
99.在一个2升的反应罐中并流加入5000毫升浓度为60克氧化铝/升、其中含核糖醇6.0克、85重量%浓磷酸8.0ml、氟化铵4g的硫酸铝溶液和浓度为6重量%的氨水溶液进行沉淀反应,反应温度为50℃,反应时间为30分钟,控制氨水溶液的流量使反应体系的ph为5.0,沉淀反应结束后,在浆液中加入适量氨水使浆液的ph值为8.7,浆液于70℃下老化120分钟后过滤,滤饼用去离子水打浆洗涤2次,滤饼经120℃干燥24小时,得到改性水合氧化铝pa1,采用xrd表征,pa1具有拟薄水铝石结构。
100.经xrd表征计算得到pa1的h值列于表1中。pa1的相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。
101.pa1经600℃焙烧4小时后得到氧化铝。用红外光谱对氧化铝表面的羟基进行测定。(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
102.(2)制备改性氧化铝载体z1:
103.取1000克上述水合氧化铝pa1和30克田菁粉(河南兰考田菁胶厂生产)并混合均匀,之后加入930毫升含20g硝酸的水溶液,在柱塞式挤条机上挤成外径为1.4mm的蝶形湿
条,然后将蝶形湿条于120℃下干燥4小时,再于600℃下焙烧3小时,得到载体z1。
104.(3)制备加氢催化剂c1:
105.取110克所述载体z1,用110毫升由钼酸铵和硝酸镍组成的混合溶液(该混合溶液中含moo
3 320克/升,nio 81克/升)浸渍该载体z1 1小时,110℃烘干4小时,400℃焙烧3小时,得到加氢催化剂c1。
106.对比例1
107.按照实施例1的方法制备拟薄水铝石dpa1、载体dz1和加氢催化剂dc1,不同的是,硫酸铝溶液中只添加浓度85重量%的磷酸8.0ml,而不含核糖醇、氟化铵,得到水合氧化铝cpa1。按照实施例1的方法采用xrd表征,cpa1具有拟薄水铝石结构,经xrd表征计算得到cpa1的h值列于表1中,相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
108.对比例2
109.按照实施例1的方法制备拟薄水铝石dpa2、载体dz2和加氢催化剂dc2,不同的是,硫酸铝溶液中不添加氟化铵,且直接控制氨水溶液的流量使反应体系的ph为8.7,沉淀反应结束后,不需要在浆液中加入氨水调节ph值,得到水合氧化铝cpa2。按照实施例1的方法采用xrd表征,cpa2具有拟薄水铝石结构,经xrd表征计算得到cpa2的h值列于表1中,相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
110.对比例3
111.按照实施例1的方法制备拟薄水铝石dpa3、载体dz3和加氢催化剂dc3,不同的是,硫酸铝溶液中只添加核糖醇6.0克,而不含浓磷酸、氟化铵,得到水合氧化铝cpa3。按照实施例1的方法采用xrd表征,cpa3具有拟薄水铝石结构,经xrd表征计算得到cpa3的h值列于表1中,相对结晶度同样列于表1。经600℃焙烧4小时后用红外光谱对氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
112.实施例2
113.该实施例用于说明本发明提供的改性拟薄水铝石、改性氧化铝和加氢催化剂及其制备方法。
114.(1)制备水合氧化铝pa2:
115.在一个2升的反应罐中并流加入4000毫升浓度为45克氧化铝/升的含85重量%浓磷酸22.1ml、硅溶胶29克、山梨糖醇4.52克/升的三氯化铝溶液和1000毫升含210克氧化铝/升、苛性系数为1.58的偏铝酸钠溶液进行沉淀反应,反应温度为80℃、调节反应物流量使得中和ph值为4.0,反应停留时间15分钟;在所得浆液中加入浓度为5重量%的稀氨水调节浆液ph至9.0,并升温至85℃,老化3小时,然后用真空过滤机进行过滤,待过滤完后,在滤饼上补充加入20升去离子水(温度85℃)冲洗滤饼约30分钟。将洗涤合格的滤饼加入到3升去离子水中搅拌成浆液,浆液用泵送入喷雾干燥器进行干燥,控制喷雾干燥器出口温度在100-110℃范围,物料干燥时间约2分钟,干燥后得到水合氧化铝pa2。按照实施例1的方法采用xrd表征,pa2具有拟薄水铝石结构,经xrd表征计算得到pa2的h值列于表1中,相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。经600℃焙烧4小时后用红外光谱对改性氧化铝
表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
116.(2)制备载体z2:
117.采用上述实施例1中步骤(2)的方法制备载体z2,不同的是,将pa2挤成ф1.5毫米的蝶形条,得到载体z2。
118.(3)制备加氢催化剂c2:
119.取100克所述载体z2,用110毫升由钼酸铵和硝酸镍组成的混合溶液(该混合溶液中含moo
3 231克/升,nio 56克/升)浸渍该载体z2 1小时,然后于120℃烘干3小时,再于420℃焙烧3小时,得到加氢催化剂c2。
120.对比例4
121.按照实施例2的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,三氯化铝溶液中不含山梨糖醇和硅溶胶,得到水合氧化铝cpa4。按照实施例1的方法采用xrd表征,cpa4具有拟薄水铝石结构,经xrd表征计算得到cpa4的h值列于表1中,相对结晶度以及p2o5的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
122.对比例5
123.按照实施例2的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,三氯化铝溶液中不含硅溶胶,且直接控制偏铝酸钠溶液的流量使反应体系的ph为9.0,沉淀反应结束后,不需要在浆液中加入氨水调节ph值,得到水合氧化铝cpa5。按照实施例1的方法采用xrd表征,cpa5具有拟薄水铝石结构,经xrd表征计算得到cpa5的h值列于表1中,相对结晶度以及p2o5的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
124.对比例6
125.按照实施例2的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,三氯化铝溶液中不含浓磷酸和硅溶胶,得到水合氧化铝cpa6。按照实施例1的方法采用xrd表征,cpa6具有拟薄水铝石结构,经xrd表征计算得到cpa6的h值列于表1中,相对结晶度同样列于表1。经600℃焙烧4小时后用红外光谱对氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
126.实施例3
127.该实施例用于说明本发明提供的改性拟薄水铝石、改性氧化铝和加氢催化剂及其制备方法。
128.(1)制备水合氧化铝pa3:
129.在一个2升的反应罐中并流加入3000毫升浓度为60克氧化铝/升、葡萄糖酸含量为4.5克/升、氢氟酸6g、含85重量%浓磷酸3.5ml的硫酸铝溶液和1000毫升含200克氧化铝/升、苛性系数为1.58的偏铝酸钠溶液进行沉淀反应,反应温度为55℃、调节反应物流量使得中和ph值为6.5,反应停留15分钟,然后在所得浆液中加入浓度为100克/升的碳酸钠溶液,调节浆液ph至9.5,并升温至75℃,老化5小时,然后用真空过滤机进行过滤,待过滤完后,在滤饼上补充加入20升去离子水(温度85℃)冲洗滤饼约30分钟。滤饼经120℃干燥24小时,得到水合氧化铝pa3。按照实施例1的方法采用xrd表征,pa3具有拟薄水铝石结构,经xrd表征计算得到pa3的h值列于表1中,相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。经
600℃焙烧4小时后用红外光谱对改性氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
130.(2)制备载体z3:
131.采用上述实施例1中步骤(2)的方法制备载体z3,不同的是,将pa3挤成ф1.6毫米的蝶形条,得到载体z3。
132.(3)制备加氢催化剂c3:
133.取100克载体z3,用110毫升由偏钨酸铵和硝酸镍组成的混合溶液(该混合溶液中含wo
3 427克/升,nio 46克/升)浸渍该载体z3 1小时,110℃烘干4小时,400℃焙烧3小时,得到加氢催化剂c3。
134.实施例4
135.按照实施例3的方法,不同的是,沉淀反应过程中,调节反应物流量使得中和ph值为7。得到水合氧化铝pa4。按照实施例1的方法采用xrd表征,pa4具有拟薄水铝石结构,经xrd表征计算得到pa4的h值列于表1中,相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。经600℃焙烧4小时后用红外光谱对改性氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
136.对比例7
137.按照实施例4的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,硫酸铝溶液中不含葡萄糖酸、氢氟酸,得到水合氧化铝cpa7。按照实施例1的方法采用xrd表征,cpa7具有拟薄水铝石结构,经xrd表征计算得到cpa7的h值列于表1中,相对结晶度以及p2o5的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
138.对比例8
139.按照实施例4的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,硫酸铝溶液中不添加氢氟酸,且直接控制偏铝酸钠溶液的流量使反应体系的ph为9.5,沉淀反应结束后,不需要在浆液中加入碳酸钠溶液调节ph值,得到水合氧化铝cpa8。按照实施例1的方法采用xrd表征,cpa8具有拟薄水铝石结构,经xrd表征计算得到cpa8的h值列于表1中,相对结晶度以及p2o5的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
140.对比例9
141.按照实施例4的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,硫酸铝溶液中不含浓磷酸、氢氟酸,得到水合氧化铝cpa9。按照实施例1的方法采用xrd表征,cpa9具有拟薄水铝石结构,经xrd表征计算得到cpa9的h值列于表1中,相对结晶度同样列于表1。经600℃焙烧4小时后用红外光谱对氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
142.实施例5
143.该实施例用于说明本发明提供的改性拟薄水铝石、改性氧化铝和加氢催化剂及其制备方法。
144.(1)制备水合氧化铝pa5:
145.在带搅拌和回流冷凝管的2升三口烧瓶中,加入异丙醇-水的共沸物(含水量为15
重量%)1000克,加入85%浓磷酸4.6ml、氧化硅20克、核糖酸15g,加入氨水调整ph为5.1,然后加热至60℃,将500克熔化的异丙醇铝通过分液漏斗缓慢滴加入烧瓶中,反应2小时后,加入氨水调节ph至8.5,回流反应20小时后,蒸出脱水异丙醇,80℃老化6小时,在老化的同时蒸出含水异丙醇,老化后的水合氧化铝过滤后,经120℃干燥24小时,得到水合氧化铝pa5。按照实施例1的方法采用xrd表征,pa5具有拟薄水铝石结构,经xrd表征计算得到pa5的h值列于表1中,相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。经600℃焙烧4小时后用红外光谱对改性氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
146.(2)将所述pa5按照实施例1的方法制备载体z5和加氢催化剂c5。
147.对比例10
148.按照实施例5的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,不向三口烧瓶中加入核糖酸和氧化硅,得到水合氧化铝cpa10。按照实施例1的方法采用xrd表征,cpa10具有拟薄水铝石结构,经xrd表征计算得到cpa10的h值列于表1中,相对结晶度以及p2o5的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
149.对比例11
150.按照实施例5的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,不向三口烧瓶中加入氧化硅,且在加入相同量的核糖酸后,接着加入氨水调节ph至8.5,然后加热至60℃,然后再将500克熔化的异丙醇铝通过分液漏斗缓慢滴加入烧瓶中,得到水合氧化铝cpa11。按照实施例1的方法采用xrd表征,cpa11具有拟薄水铝石结构,经xrd表征计算得到cpa11的h值列于表1中,相对结晶度以及p2o5的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
151.对比例12
152.按照实施例5的方法制备拟薄水铝石、载体和加氢催化剂,不同的是,不向三口烧瓶中加入浓磷酸和氧化硅,得到水合氧化铝cpa12。按照实施例1的方法采用xrd表征,cpa12具有拟薄水铝石结构,经xrd表征计算得到cpa12的h值列于表1中,相对结晶度同样列于表1。经600℃焙烧4小时后用红外光谱对氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
153.实施例6
154.该实施例用于说明本发明提供的改性拟薄水铝石、改性氧化铝和加氢催化剂及其制备方法。
155.在带搅拌和回流冷凝管的2升三口烧瓶中,加入异丙醇-水的共沸物(含水量为15重量%)1000克,加入85%浓磷酸7.0ml,氟化铵20g,核糖酸12g,加入氨水调整ph至6.2,加热至60℃,将500克熔化的异丙醇铝通过分液漏斗缓慢滴加入烧瓶中,反应5小时后,加入氨水调节ph至8.5,回流反应20小时后,蒸出脱水异丙醇,80℃老化6小时,在老化的同时蒸出含水异丙醇,老化后的水合氧化铝过滤后,经120℃干燥24小时,得到水合氧化铝pa6。按照实施例1的方法采用xrd表征,pa6具有拟薄水铝石结构,经xrd表征计算得到pa6的h值列于表1中,相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。经600℃焙烧4小时后用红外光谱对氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
156.将所述pa6按照实施例1的方法制备载体z6和加氢催化剂c6。
157.对比例13
158.按《重油加氢催化剂载体材料的研究》中的典型方法制备含磷拟薄水铝石,用添加85%的浓磷酸8.8ml浓度为57g
·
l-1
的硫酸铝溶液3000ml,与浓度为64g
·
l-1
偏铝酸钠溶液2500ml进行沉淀反应,中和ph为8.0,反应时间为70min,然后进行老化,老化温度为90℃,老化ph为8.5,老化后过滤,滤饼用去离子水打浆洗涤2次,滤饼经120℃干燥24小时制备得到含磷拟薄水铝石cpa13。按照实施例1的方法采用xrd表征,cpa13具有拟薄水铝石结构,经xrd表征计算得到cpa13的h值列于表1中,相对结晶度以及p2o5的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
159.将所述cpa13按照实施例1的方法制备得到dz13和dc13。
160.对比例14
161.按cn103721732a中公开了一种加磷改性拟薄水铝石催化剂载体材料及制备方法。在中和反应釜1中,加入氧化铝浓度50g/l的硫酸铝溶液和氧化铝浓度为220g/l、苛性比为1.2的偏铝酸钠溶液,控制ph为7.0,温度为55℃;中和反应釜1的浆液通过溢流反应管流入中和反应釜2,同时向中和反应釜2中加入浓度为150g/l的碳酸钠溶液,控制ph为9.5,控制反应温度在70℃;中和反应釜2的浆液通过溢流反应管流入老化反应釜,老化反应釜内浆液温度95℃,老化2小时;根据中和反应釜1反应过程中投入氧化铝的质量,计算向老化反应釜加入五氧化二磷浓度为100g/l的磷酸溶液的体积,加入磷酸的五氧化二磷含量为氧化铝含量的4%;老化完成后洗涤、干燥,得到含磷拟薄水铝石。按照实施例1的方法采用xrd表征,cpa14具有拟薄水铝石结构,经xrd表征计算得到cpa14的h值列于表1中,相对结晶度同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
162.将所述cpa14按照实施例1的方法制备得到dz14和dc14。
163.实施例7
164.按照实施例1的方法制备改性拟薄水铝石、改性氧化铝和加氢催化剂,不同的是,在硫酸铝溶液中还加入硅溶胶17g,得到水合氧化铝pa7。按照实施例1的方法采用xrd表征,pa8具有拟薄水铝石结构,经xrd表征计算得到pa7的h值列于表1中,相对结晶度以及p2o5以及非金属助剂的含量同样列于表1。经600℃焙烧4小时后用红外光谱对含磷氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
165.实施例8
166.按照实施例5的方法制备改性拟薄水铝石、改性氧化铝和加氢催化剂,不同的是,不向三口烧瓶中加入氧化硅,而加入氟化铵15g,得到改性拟薄水铝石pa8。
167.按照实施例1的方法采用xrd表征,pa8具有拟薄水铝石结构,经xrd表征计算得到pa10的h值列于表1中,相对结晶度同样列于表1。经600℃焙烧4小时后用红外光谱对氧化铝表面的羟基进行测定,(i
3670
i
3580
)/(i
3770
i
3720
)的值列于表1中。
168.表1
[0169][0170][0171]
注:m表示(i
3670
i
3580
)/(i
3770
i
3720
)的值
[0172]
从表1的结果可以看出,采用本发明提供的方法制备的改性拟薄水铝石具有1.7≤h≤4的特征,优选2.2≤h≤3.5,而采用现有技术的方法以及对比例中方法制备的各种拟薄水铝石的h值均小于1.7。从表1的结果可以看出,采用本发明方法制备的改性拟薄水铝石经600℃焙烧后得到改性氧化铝的ir表征谱图中,羟基具有特征(i
3670
i
3580
)/(i
3770
i
3720
)为1.9-3.5,优选2-3.3,而采用现有技术方法以及对比例中方法制备的拟薄水铝石,经600℃焙烧后得到氧化铝的ir表征谱图中,羟基特征(i
3670
i
3580
)/(i
3770
i
3720
)<1.8。
[0173]
测试例1
[0174]
以一种劣质重油(硫元素含量为2.5重量%、氮元素含量为0.52重量%、ni含量32μg/g、v含量24μg/g、残炭值为9.7重量%的劣质渣油)为原料,在100毫升小型固定床反应器
上评价催化剂。
[0175]
分别将上述100ml实施例1-8以及对比例1-14对应制得的加氢催化剂破碎成直径2-3毫米的颗粒后进行预硫化,预硫化条件包括:硫化油采用含5w%二甲基二硫醚的青岛常一线煤油,硫化油的液时体积空速1.2h-1
,氢分压14.0mpa,氢油体积比600,在360℃恒温硫化3小时。然后在反应温度380℃、氢分压15兆帕、液时空速为0.6小时-1
,氢油体积比为600的条件下反应100小时后取样分析,催化剂装填量为100毫升,以评价催化剂的加氢活性和稳定性,结果如表2所示。
[0176]
其中,所述脱(ni v)率、脱硫率和脱残炭率的计算方法相同;本发明以脱(ni v)率为例进行示例性说明计算方法,脱(ni v)率=(原料中(ni v)含量-加氢后产品中(ni v)含量)/原料中(ni v)含量。
[0177]
其中,油样中镍和钒的含量采用电感耦合等离子体发射光谱仪(icp-aes)测定(所用仪器为美国pe公司pe-5300型等离子体光量计,具体方法见石油化工分析方法ripp124-90);
[0178]
油样中硫含量使用电量法测定(具体方法见石油化工分析方法ripp62-90);
[0179]
油样中残炭含量使用微量法测定(具体方法见石油化工分析方法ripp149-90)。
[0180]
表2
[0181]
[0182]
从表2可以看出,采用本发明提供的改性拟薄水铝石焙烧后制得的改性氧化铝用作加氢催化剂载体时,该催化剂在其它条件相同的情况下具有更好的加氢活性,也能看出同时具有很好的稳定性。
[0183]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。