1.本发明涉及有机高分子材料领域,具体涉及一种聚酰亚胺及其制备方法。
背景技术:
2.聚酰亚胺(polyimide,pi)是指主链上含有酰亚胺环(
‑
co
‑
n
‑
co
‑
)的一类聚合物。聚酰亚胺被认为是综合性能最佳的有机高分子材料之一,它的耐热温度可以达到400℃以上,具备很高的绝缘性能,介电常数与介电损耗很小,并且具备良好的化学稳定性。总之,得益于聚酰亚胺具备的十分出色的热稳定性、化学稳定性、力学性能和独特的电性能等,其在许多领域都展现了很好的应用潜力。但是,由于聚酰亚胺不易溶解或熔融,因此材料加工性差。为改善其加工性能,可采用两步法制备聚酰亚胺,即,首先通过聚合单体制备聚酰胺酸paa(聚酰亚胺前体溶液),可在其中添加基本上不会增加聚酰胺酸溶液的粘度(即,基本上不参与分子链增长)的四羧酸作为粘度调节剂以增加其加工性能;然后,再通过热法(直接加热)或者化学法(添加脱水剂,催化剂等)进一步脱水环化制备得聚酰亚胺。图1为聚酰亚胺的合成方法的示意图,以ppd和bpda作为聚合单体,dmac为溶剂为例。
3.研究表明,聚酰胺酸的环化缩聚反应从120℃开始,在170℃附近到达最大的反应速率,到250℃反应基本结束。然而,用上述方法制备聚酰亚胺膜时(以nmp,即n
‑
甲基吡咯烷酮作为溶剂为例),聚酰胺酸涂覆在载体上之后,需通过真空干燥箱真空干燥,一般地,经过此操作后,约80%的nmp溶剂已经挥发。进一步地,采用热法固化过程中,当温度升至200℃(nmp的沸点204℃)左右时,nmp基本已挥发完全,此时的固化反应(失水环化反应)接近于固相反应,分子链运动能力低,不利于聚酰亚胺分子链的增长,进而降低了聚酰亚胺膜的性能,尤其是机械性能(主要指拉伸强度,拉伸模量和拉伸伸长率)。
4.另一方面,高温固化过程中,采用缓慢梯度升温有利于nmp的缓慢挥发,进而有利于获得表面平整的聚酰亚胺膜;并可在一定程度上保持聚酰亚胺分子链的面内取向效应,进而获得较低cte的聚酰亚胺膜。但是,缓慢梯度升温在实际工业生产应用中,会降低生产效率,增大能源消耗,提高生产成本。
5.郑森森等(郑森森,郭涛,董杰,王士华,张清华.含咪唑结构高强高模聚酰亚胺纤维的制备及其结构与性能[j].纺织学报,2021,42(02):7
‑
11 20.doi:10.13475/j.fzxb.20200708006)公开了一种含咪唑结构的二胺单5
‑
氨基
‑2‑
(对氨基苯基)苯并咪唑引入bpda和ppd分子体系中,通过干法纺丝工艺制备力学性能优异的聚酰亚胺纤维。cn 109535423 a公开了通过添加咪唑化合物获得机械特性优异的透明的聚酰亚胺的制造方法。但是,上述方法均不能有效实现缩短固化时间的同时保持聚酰亚胺优异的机械性能。
技术实现要素:
[0006]
为解决以上至少之一的技术问题,本发明的目的在于提供一种聚酰亚胺及其制备方法,通过添加含有烷氧基的羧酸衍生物制备具有良好加工性能的聚酰胺酸清漆,并可通过固化过程获得具有优异的热稳定性及机械性能的聚酰亚胺材料。
[0007]
为了实现以上目的,本发明采用以下技术方案:
[0008]
本发明一方面提供一种聚酰亚胺的制备方法,包括以下步骤:
[0009]
使二胺和四羧酸二酐在溶剂中发生聚合反应生成聚酰胺酸;
[0010]
加入含有烷氧基的羧酸衍生物进行反应,得到聚酰亚胺酸清漆;
[0011]
将所述聚酰亚胺酸清漆固化得到聚酰亚胺;
[0012]
其中,所述含有烷氧基的羧酸衍生物为四羧酸二酐化合物水解开环以后,两个羧基中的羟基基团分别被两个烷氧基取代的化合物,且上述的两个羧基来自不同的酸酐基团。所述含有烷氧基的羧酸衍生物的通式可表示为式(1):
[0013][0014]
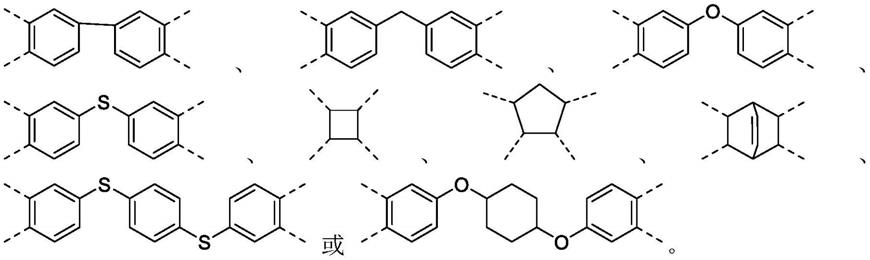
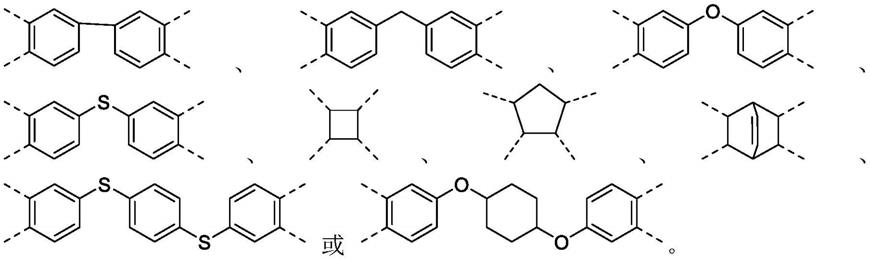
式(1)中,x为具有芳香环结构或者脂肪环结构的四价基团,r1和r2各自独立地选自烷氧基;对所述烷氧基部分的碳原子个数没有特别限制,优选碳原子个数为1~3,r1和r2的具体结构可分别为,但不限于:
‑
och3、
‑
och2ch3、
‑
och2ch2ch3、
‑
och(ch3)ch3。
[0015]
根据本发明聚酰亚胺的制备方法,优选地,x选自环碳原子数为4
‑
8的脂肪环结构的四价基团或
[0016]
a选自单键、烷基、o、s或其中,b环选自脂肪环或苯环,z选自o或s。
[0017]
根据本发明聚酰亚胺的制备方法,优选地,所述b环为环己烷或苯环。
[0018]
根据本发明聚酰亚胺的制备方法,优选地,所述b环为对位取代。
[0019]
根据本发明聚酰亚胺的制备方法,优选地,x的结构选自:
[0020][0021]
本领域技术人员理解的,以上结构式中的虚线代表该基团与其他基团之间的连接键。
[0022]
根据本发明聚酰亚胺的制备方法,优选地,所述二胺选自但不限于对苯二胺(para
‑
phenylenediamine,ppd)、间苯二胺(m
‑
phenylenediamine,mpd)、4,4
’‑
二氨基二苯醚(4,4'
‑
oxydianiline,oda)、对亚甲基二苯胺(para
‑
methylene dianiline,pmda)、间亚甲基二苯胺(meta
‑
methylene dianiline,mmda)、1,3
‑
双(3
‑
氨基苯氧基)苯(1,3
‑
bis(3
‑
aminophenoxy)benzene,133apb)、1,3
‑
双(4
‑
氨基苯氧基)苯(1,3
‑
bis(4
‑
aminophenoxy)benzene,134apb)、双(三氟甲基)联苯胺(tfdb)、2,2
’‑
双[4(4
‑
氨基苯氧基)苯基]六氟丙烷(2,2'
‑
bis[4(4
‑
aminophenoxy)phenyl]hexafluoropropane,4bdaf)、2,2
’‑
双(3
‑
氨基苯基)六氟丙烷(2,2'
‑
bis(3
‑
aminophenyl)hexafluoropropane,33
‑
6f)、2,2
’‑
双(4
‑
氨基苯基)六氟丙烷(2,2'
‑
bis(4
‑
aminophenyl)hexafluoropropane,44
‑
6f)、双(4
‑
氨基苯基)砜(bis(4
‑
aminophenyl)sulfone,4dds)、双(3
‑
氨基苯基)砜(bis(3
‑
aminophenyl)sulfone,3dds)、1,3
‑
环己烷二胺(1,3
‑
cyclohexanediamine,13chd)、1,4
‑
环己烷二胺(1,4
‑
cyclohexanediamine,14chd)、2,2
’‑
双[4
‑
(4
‑
氨基苯氧基苯基)]丙烷(2,2
‑
bis[4
‑
(4
‑
aminophenoxy)
‑
phenyl]propane,6hmda)、2,2
‑
双(3
‑
氨基
‑4‑
羟基苯基)六氟丙烷2,2
‑
bis(3
‑
amino
‑4‑
hydroxy
‑
phenyl)
‑
hexafluoropropane,dboh、4,4
’‑
双(3
‑
氨基苯氧基)二苯基砜(4,4'
‑
bis(3
‑
amino phenoxy)diphenyl sulfone,dbsda)、9,9
‑
双(4
‑
氨基苯基)芴(9,9
‑
bis(4
‑
aminophenyl)fluorene,fda)、9,9
‑
双(3
‑
氟
‑4‑
氨基苯基)芴(9,9
‑
bis(3
‑
fluoro
‑4‑
aminophenyl)fluorene,ffda)中的一种或两种以上的组合。
[0023]
根据本发明聚酰亚胺的制备方法,优选地,所述四羧酸二酐选自但不限于3,3',4,4'
‑
联苯四羧酸二酐(bpda)、均苯四甲酸二酐(pmda)、2,2
‑
双(3,4
‑
二羧基苯基)六氟丙烷二酐(6fda)、4
‑
(2,5
‑
二氧四氢呋喃
‑3‑
基)
‑
1,2,3,4
‑
四氢化萘
‑
1,2
‑
二羧酸酐(tda)、3,3,4,4
‑
二苯甲酮四羧酸二酐(3,3',4,4'
‑
benzophenone tetracarboxylic dianhydride,btda)、4,4
’‑
氧二邻苯二甲酸二酐(4,4
‑
oxydiphthalic dianhydride,odpa)、双(3,4
‑
二羧基苯基)二甲基硅烷二酐(bis(3,4
‑
dicarboxyphenyl)dimethyl
‑
silane dianhydride,sida)、4,4
‑
双(3,4
‑
二羧基苯氧基)二苯硫醚二酐(4,4
‑
bis(3,4
‑
dicarboxyphenoxy)diphenyl sulfide dianhydride,bdsda),磺酰基二邻苯二甲酸酐(sulfonyldiphthalic anhydride,so2dpa)、环丁烷四甲酸二酐(cyclobutane
‑
1,2,3,4
‑
tetracarboxylic dianhydride,cbda)、4,4
’‑
(4,4
’‑
异丙基二苯氧基)双(邻苯二甲酸酐)(4,4'
‑
(4,4'
‑
isopropylidenediphenoxy)bis(phthalicanhydride),6hbda)中的一种或两种以上的组合。
[0024]
根据本发明聚酰亚胺的制备方法,优选地,所述溶剂选自但不限于n
‑
甲基吡咯烷酮(nmp)、二甲基甲酰胺(dmf)、二甲基乙酰胺(dmac)、二甲基亚砜(dmso)、丙酮、乙酸乙酯、二乙基甲酰胺(def)、二乙基乙酰胺(dea)、丙二醇甲醚(propylene glycol monomethyl ether,pgme)、m
‑
甲酚等极性溶剂中的一种或两种以上的混合溶剂。此外,也可以使用四氢呋喃(thf)、氯仿等低沸点溶液或γ
‑
丁内酯等低吸收性溶剂。更优选地,所述溶剂为二甲基乙酰胺(dmac)、n
‑
甲基吡咯烷酮或二者的混合溶剂。
[0025]
基于本发明聚酰亚胺的制备方法,通过使作为四羧酸成分的四羧酸二酐与二胺成分以基本上等摩尔量(优选地,所述二胺和四羧酸二酐的摩尔比为0.90
‑
1.10,更优选为0.95
‑
1.05),在溶剂中于例如120℃以下的相对较低温度反应,同时抑制酰亚胺化,可以获得聚酰胺酸清漆。对溶剂量的要求是从生成的聚酰胺酸清漆的固体质量分数来限定的,优选地,所述聚酰亚胺酸清漆的固体质量分数为5%
‑
50%,更优选10%
‑
25%。
[0026]
另外,当二胺以四羧酸二酐与二胺等摩尔计过量时,必要时可以以基本上对应于二胺成分的过量摩尔数的量添加所述含有烷氧基的羧酸衍生物,所述含有烷氧基的羧酸衍生物与二胺的摩尔比可为(0.001
‑
0.5):2,优选(0.01
‑
0.1):1,从而使得反应过程中的四羧
酸二酐(所述含有烷氧基的羧酸衍生物在加热过程中会转化为四羧酸二酐)与二胺的摩尔比更接近于基本上等摩尔量。即:所述含有烷氧基的羧酸衍生物与二胺的摩尔比可为(0.001
‑
0.5):2,优选为(0.01
‑
0.1):1;所述含有烷氧基的羧酸衍生物与所述四羧酸二酐的总和与所述二胺的摩尔比为1:(0.90
‑
1.10),优选为1:(0.95
‑
1.05),更优选为1:1。
[0027]
作为本发明所用的含有烷氧基的羧酸衍生物,优选基本上不会增加聚酰胺酸清漆粘度(即,基本上不参与分子链增长)的四羧酸衍生物。理论上,如式(1)所示的含有烷氧基的羧酸衍生物均可以作为原料,在加热过程中转化为四羧酸二酐,进而与二胺反应生成聚酰胺酸,例如本发明具体实施方式中所使用的mabm转化为四羧酸二酐的温度为130℃,mabe转化为四羧酸二酐的温度为160℃;而s
‑
bpta转化为四羧酸二酐的温度为200℃(如图2所示)。但是考虑实际应用中,含有烷氧基的羧酸衍生物在加热转化为四羧酸二酐的过程中会脱去醇类小分子,该醇类小分子的沸点也会影响聚酰胺酸以及之后固化的聚酰亚胺的性质。因此,所述含有烷氧基的羧酸衍生物优选为:
[0028][0029]
更具体地,所述聚酰胺酸的制备过程具体包括:
[0030]
将二胺溶解于有机溶剂中,向其中逐渐添加四羧酸二酐,并同时搅拌溶液,之后在0℃至120℃(优选20℃至80℃)的温度下继续搅拌1
‑
72小时;但制备方法不限于此。
[0031]
当其在80℃以上的温度反应时,产物聚酰胺酸的分子量可以根据聚合时的温度而变化,并且酰亚胺化可以借助热而进行,因此可能无法稳定地制备聚酰亚胺前体。上述的制备方法中的二胺和四羧酸二酐的添加顺序是优选的,因为聚酰胺酸的分子量容易增加(因为四羧酸二酐在以上溶剂中的溶解性十分有限,但二胺在其中的溶解性优良,生成的聚酰胺酸清漆在其中也具有良好的溶解性),且该顺序同时因沉淀量减少(所述沉淀为不能完全溶解在溶剂中的四羧酸二酐)而优选。此外,上述制备方法中的二胺和四羧酸二酐的添加顺序也可以颠倒。
[0032]
根据本发明聚酰亚胺的制备方法,步骤“将所述聚酰亚胺酸清漆固化得到聚酰亚胺”中,首先将所述聚酰亚胺酸清漆通过涂覆、静电纺丝等方式制备成出所需形态的涂膜,之后在n2气氛中通过热法(梯度升温)固化成聚酰亚胺。优选地,所述热法固化为梯度升温;更优选地,所述梯度升温的升温速率为2
‑
10℃/min,且流程包括:升温至100℃保持0.5
‑
1h,升温至150℃保持0.5
‑
1h,升温至200℃保持0.5
‑
1h,升温至250℃保持0.5
‑
1h,升温至300℃保持0.5
‑
1h,升温至350℃保持0.5
‑
1h,每个升温阶段的升温速率可以相同也可以不同。
[0033]
例如在一优选实施方案中,所述梯度升温的升温速率为4
‑
5℃/min,且流程包括:升温至100℃保持1h,升温至150℃保持1h,升温至200℃保持0.5h,升温至250℃保持0.5h,升温至300℃保持0.5h,升温至350℃保持0.5h。
[0034]
对聚酰胺酸清漆的涂膜进行固化,使聚酰胺酸进行酰亚胺化(即闭环)。具体方法为热法,即在高温条件下脱水环化。本发明对于具体固化过程中使用的添加剂及升温过程无特别限制。
[0035]
本发明通过添加含有烷氧基的羧酸衍生物,可制备具有良好加工性能的聚酰胺酸清漆,并可通过固化过程获得具有优异的热稳定性及机械性能的聚酰亚胺材料。同时,通过本发明提供的方法还可缩短固化时间,提高生产效率。本发明得到的聚酰亚胺膜可以适合用于显示器、触摸屏或太阳能电池的基板。
[0036]
本发明另一方面提供一种由以上制备方法获得的聚酰亚胺;优选地,所述聚酰亚胺为聚酰亚胺膜。
[0037]
本发明与现有技术相比具有以下有益效果:
[0038]
1)本发明通过添加含有烷氧基的羧酸衍生物作为粘度调节剂,在聚酰胺酸聚合环化的过程中,反应物与产物的体系能够降低粘度调节剂转化为反应物(四羧酸二酐)的温度,使得粘度调节剂转化为反应物参与反应,有利于聚合物分子链的增长,进而提高聚酰亚胺制成的薄膜的机械性能(如拉伸强度和伸长率)。
[0039]
2)缩短了聚酰胺酸清漆固化成聚酰亚胺的时间,提高了生产效率,降低了反应消耗能量,从而节约了生产成本。
[0040]
3)相较于四羧酸化合物,所述含有烷氧基的羧酸衍生物的添加可使固化反应在较低温度趋向完全,提升了所述含有烷氧基的羧酸衍生物在加热过程中生成的四羧酸二酐与二胺的反应效率,降低了因高温环境溶剂大量挥发接近固相反应的固化反应中未参与反应的四羧酸二酐的量,从而降低了聚酰亚胺膜在高温下的释气现象。
附图说明
[0041]
图1为现有技术中聚酰亚胺的合成方法示意图(以ppd和bpda作为聚合单体为例)。
[0042]
图2为本发明实施例及对比例中所使用粘度调节剂转化为反应物的反应过程示意图。
具体实施方式
[0043]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0044]
本发明所有数值指定(例如温度、时间、浓度及重量等,包括其中每一者的范围)通常可是适当以0.1或1.0的增量改变( )或(
‑
)的近似值。所有数值指定均可理解为前面有术语“约”。
[0045]
其中,四羧酸二酐与二胺的简称分别如下所述:
[0046]
bpda:3,3',4,4'
‑
联苯四甲酸二酐
[0047][0048]
pmda:均苯四甲酸二酐(对应含有烷氧基的羧酸衍生物缩写为cmob)
[0049][0050]
4,4'
‑
oda:4,4'
‑
二氨基二苯醚
[0051][0052]
tfdb:双(三氟甲基)联苯胺
[0053][0054]
ppd:对苯二胺;
[0055]
s
‑
bpta:3,3',4,4'
‑
联苯四甲酸
[0056][0057]
nmp:n
‑
甲基吡咯烷酮;
[0058]
含有烷氧基的羧酸衍生物:
[0059]
1)mabm,mabm结构式如下:
[0060][0061]
2)mabe,mabe结构式如下:
[0062][0063]
对比例1a
[0064]
n2气氛下,先将1.00mol ppd加入到2196.7g的nmp中搅拌至ppd完全溶解,再向反应器中加入0.95mol的bpda于50℃下使用机械搅拌反应5h,随后加入0.05mol的s
‑
bpta,搅拌混合均匀后可获得聚酰胺酸(paa)溶液(即聚酰胺酸清漆)。
[0065]
将paa溶液使用涂布机在玻璃上涂覆成膜。再将其置于n2气氛中,升温速率为5℃/min,按100℃/1h
→
150℃/1h
→
200℃/0.5h
→
250℃/0.5h
→
300℃/0.5h
→
350℃/0.5h的程序进行热亚胺化,得到聚酰亚胺薄膜,膜厚为10μm。
[0066]
对比例1b
[0067]
n2气氛下,先将1.00mol ppd加入到2196.7g的nmp中搅拌至ppd完全溶解,再向反应器中加入0.95mol的bpda于50℃下使用机械搅拌反应5h,随后加入0.05mol的s
‑
bpta,搅拌混合均匀后可获得聚酰胺酸(paa)溶液。
[0068]
将paa溶液使用涂布机在玻璃上涂覆成膜。再将其置于n2气氛中,升温速率为5℃/min,按100℃/1h
→
150℃/1h
→
200℃/1h
→
250℃/0.5h
→
300℃/0.5h
→
350℃/0.5h的程序进行热亚胺化,得到聚酰亚胺薄膜,膜厚平为10μm。
[0069]
实施例2
[0070]
n2气氛下,先将1.00mol ppd加入到2196.7g的nmp中搅拌至ppd完全溶解,再向反应器中加入0.95mol的bpda于50℃下使用机械搅拌反应5h,随后加入0.05mol的mabm,搅拌混合均匀后可获得聚酰胺酸(paa)溶液。
[0071]
将paa溶液使用涂布机在玻璃上涂覆成膜。再将其置于n2气氛中,升温速率为5℃/min,按100℃/1h
→
150℃/1h
→
200℃/0.5h
→
250℃/0.5h
→
300℃/0.5h
→
350℃/0.5h的程序进行热亚胺化,得到聚酰亚胺薄膜,膜厚为10μm。
[0072]
实施例3
[0073]
n2气氛下,先将1.00mol ppd加入到2196.7g的nmp中搅拌至ppd完全溶解,再向反应器中加入0.95mol的bpda于50℃下使用机械搅拌反应5h,随后加入0.05mol的mabe,搅拌混合均匀后可获得聚酰胺酸(paa)溶液。
[0074]
将paa溶液使用涂布机在玻璃上涂覆成膜。再将其置于n2气氛中,升温速率为5℃/min,按100℃/1h
→
150℃/1h
→
200℃/1h
→
250℃/0.5h
→
300℃/0.5h
→
350℃/0.5h的程序进行热亚胺化,得到聚酰亚胺薄膜,膜厚为10μm。
[0075]
实施例4
[0076]
n2气氛下,先将1.00mol tfdb加入到2196.7g的nmp中搅拌至tfdb完全溶解,再向反应器中加入0.98mol的bpda于50℃下使用机械搅拌反应5h,随后加入0.05mol的mabm,搅拌混合均匀后可获得聚酰胺酸(paa)溶液。
[0077]
将paa溶液使用涂布机在玻璃上涂覆成膜。再将其置于n2气氛中,升温速率为5℃/min,按100℃/1h
→
150℃/1h
→
200℃/1h
→
250℃/0.5h
→
300℃/0.5h
→
350℃/0.5h的程序进行热亚胺化,得到聚酰亚胺薄膜,膜厚为10μm。
[0078]
实施例5
[0079]
n2气氛下,先将1.00mol ppd加入到2196.7g的nmp中搅拌至ppd完全溶解,再在反应器中加入0.94mol的pmda于50℃下使用机械搅拌反应5h,随后加入0.08
‑
mol的cmob,搅拌混合均匀后可获得聚酰胺酸(paa)溶液。
[0080]
将paa溶液使用涂布机在玻璃上涂覆成膜。再将其置于n2气氛中,升温速率为5℃/min,按100℃/1h
→
150℃/1h
→
200℃/0.5h
→
250℃/0.5h
→
300℃/0.5h
→
350℃/0.5h的程序进行热亚胺化,得到聚酰亚胺薄膜,膜厚为10μm。
[0081]
实施例6
[0082]
n2气氛下,先将1.00mol 4,4'
‑
oda加入到2196.7g的dmac中搅拌至4,4'
‑
oda完全溶解,再在反应器中加入0.95mol的pmda于55℃下使用机械搅拌反应5h,随后加入0.07mol的cmob,搅拌混合均匀后可获得聚酰胺酸(paa)溶液。
[0083]
将paa溶液使用涂布机在玻璃上涂覆成膜。再将其置于n2气氛中,升温速率为5℃/min,按100℃/0.5h
→
150℃/1h
→
200℃/0.5h
→
250℃/1h
→
300℃/0.5h
→
350℃/0.5h的程序进行热亚胺化,得到聚酰亚胺薄膜,膜厚为10μm。
[0084]
机械性能测试
[0085]
根据标准方法“astm d882”,使用万能材料试验机(ez
‑
lx 5000n,日本岛津)测试薄膜的机械力学性能,包括拉伸强度、拉伸模量以及拉伸伸长率,室温下拉伸速率10mm/min。
[0086]
释气测试(outgas)
[0087]
使用热重分析仪(dtg
‑
60,日本岛津)测试薄膜的释气,具体测试条件为:氮气氛围中,25℃升温至450℃,再在450℃温度下保温2h,升温速率为10℃/min。
[0088]
以上性能测试结果如下表1所示:
[0089]
表1
[0090][0091]
对比例1a和1b使用相同原料配比,但是对比例1b的固化升温时间在200℃时采用保温1h,比对比例1a对应的保温时间长0.5h。如前文所述,200℃上下的温度阶段对应溶剂nmp的强烈挥发、s
‑
bpta的脱水酸酐化以及聚酰胺酸的脱水环化反应,固化时间更长的对比例1b表现出较好的机械性能。
[0092]
另外,对比低比例1a和实施例2中的聚酰亚胺薄膜样品可以看出,在相同实验条件及固化温度梯度的条件下,实施例2制备出的聚酰亚胺薄膜的机械性能(具体指拉伸强度,拉伸模量以及拉伸伸长率)更加优良,效果明显。
[0093]
同样地,对比对比例1b和实施例3中的聚酰亚胺薄膜样品可以看出,实施例3制备所得的聚酰亚胺薄膜的机械性能更加优良。
[0094]
对比实施例2
‑
6,实施例2可实现在固化时间较短的条件下生成的聚酰亚胺薄膜的综合性能最为优良。另外,实施例3中所需固化时间较长,但制备出的酰亚胺薄膜的综合性能与对比例1b相比仍较为优良。实施例4
‑
6表明,使用不同的二胺、二酐、对应的含有烷氧基的羧酸衍生物和溶剂,同样能获得性能较优的聚酰亚胺薄膜。
[0095]
通过以上对比例及实施例可以看出,含有烷氧基的羧酸衍生物(mabm、mabe)的添
加可以实现在缩短聚酰胺酸加热固化的时间的同时提升聚酰亚胺材料的机械性能。
[0096]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。