用于制备取代多环吡啶酮衍生物及其晶体的方法
1.本发明申请是基于申请日为2017年06月19日,申请号为201780038096.2(国际申请号为pct/jp2017/022478)、名称为“用于制备取代多环吡啶酮衍生物及其晶体的方法”的发明专利申请的分案申请。
[技术领域]
[0002]
本发明涉及用于制备取代多环吡啶酮衍生物及其晶体的方法。具体地,本发明涉及用于制备具有帽依赖性(cap
‑
dependent)核酸内切酶抑制活性的取代多环吡啶酮衍生物及其中间体的方法。
[0003]
[发明背景]
[0004]
wo 2010/110409(专利文献1)公开了一种用于使用吡喃酮衍生物和吡啶酮衍生物制备多环吡啶酮衍生物的方法(实施例3)。
[0005]
[化学方案1]
[0006][0007]
wo 2010/147068(专利文献2)和wo 2012/039414(专利文献3)公开了一种使用吡啶酮衍生物制备多环吡啶酮衍生物的方法(实施例165)。
[0008]
[化学方案2]
[0009][0010]
然而,专利文献1至3没有描述在光学活性多环吡啶酮衍生物与硫杂卓(thiepin)衍生物的偶联步骤中使用苄基保护的多环吡啶酮衍生物降低了产物的光学纯度。而且,在专利文献1至3中既没有描述也没有暗示当使用己基保护的多环吡啶酮衍生物进行偶联反应时,该偶联反应以良好的产率进行而不降低光学纯度。此外,既没有描述也没有暗示当在反应中存在镁盐下进行反应以将多环吡啶酮衍生物中的保护基团从不同于未取代烷基的保护基团交换成为未取代烷基时,该反应以高产率进行而不降低光学纯度。
[0011]
专利文献1公开了以下方法,其包括使苄基保护的多环吡啶酮衍生物与二苯甲基衍生物偶联的步骤(实施例21)。然而,既没有描述也没有暗示对多环吡啶酮衍生物中的保护基团进行交换的任何步骤。
[0012]
[化学方案3]
[0013][0014]
专利文献2公开了使取代三环吡啶酮衍生物与二苯甲基衍生物偶联的步骤(实施例175)。然而,既没有描述也没有暗示对三环吡啶酮衍生物中的保护基团进行交换的任何步骤。
[0015]
[化学方案4]
[0016][0017]
专利文献2公开了使取代三环吡啶酮衍生物与硫杂卓衍生物偶联的步骤(实施例583和584)。然而,既没有描述也没有暗示对三环吡啶酮衍生物中的保护基团进行交换的任何步骤或光学纯度的降低。
[0018]
[化学方案5]
[0019][0020]
[现有技术文献]
[0021]
[专利文献]
[0022]
wo 2010/110409
[0023]
wo 2010/147068
[0024]
wo 2012/039414
[0025]
[发明概述]
[0026]
[技术问题]
[0027]
pct/jp2016/63139描述了该化合物,其为本文公开的具有式(v)或(vi)的化合物,
具有帽依赖性核酸内切酶抑制活性且可用作用于由感染流感病毒所造成的症状和/或疾病的治疗剂和/或预防剂。
[0028]
本发明的一个目的是提供一种用于制备具有帽依赖性核酸内切酶抑制活性的具有式(v)或(vi)的取代多环吡啶酮衍生物及其具有式(ii)或(iv)的中间体的新颖且有用的方法。
[0029]
[问题的解决方案]
[0030]
诸位发明人已发现,在光学活性的取代三环吡啶酮衍生物与硫杂卓衍生物的偶联步骤中发生光学活性的取代环状吡啶酮衍生物的光学纯度降低。
[0031]
诸位发明人还已发现一种通过从不同于未取代烷基的保护基团(例如苄基)交换成己基来实现光学活性的取代三环吡啶酮衍生物与硫杂卓衍生物的偶联反应而不降低光学纯度的方法。
[0032]
本发明涉及以下内容。
[0033]
(1)一种用于制备具有式(ii)的化合物的方法:
[0034]
[化学方案6]
[0035][0036]
其中r2是未取代烷基;
[0037]
其特征在于,在钠盐和/或镁盐的存在下使具有式(i)的化合物:
[0038]
[化学方案7]
[0039][0040]
其中r1是氢或不同于未取代烷基的保护基团,
[0041]
与具有式r2‑
oh的化合物反应,其中r2是如上所定义的。
[0042]
(2)根据(1)的方法,其中该反应在镁盐的存在下进行。
[0043]
(3)根据(1)的方法,其中该反应在异丙基氯化镁的存在下进行。
[0044]
(4)根据(1)至(3)中任一项的方法,其中r1是苄基。
[0045]
(5)根据(1)至(4)中任一项的方法,其中r2是己基。
[0046]
(6)一种用于制备具有式(iv)的化合物的方法:
[0047]
[化学方案8]
[0048][0049]
其中r3、r4、r5和r6各自独立地是氢或卤素,条件是r3、r4、r5和r6中的一个或两个是卤素,
[0050]
其特征在于,使具有式(ii')的化合物:
[0051]
[化学方案9]
[0052][0053]
与具有式(iii)的化合物反应:
[0054]
[化学方案10]
[0055][0056]
其中r3、r4、r5和r6是如上所定义的。
[0057]
(7)根据(6)的方法,其中r3是氢,r4是氢,r5是氟,并且r6是氟。
[0058]
(8)一种用于制备具有式(v)或式(vi)的化合物的方法:
[0059]
[化学方案11]
[0060][0061]
该方法包括根据(1)至(7)中任一项的方法。
[0062]
(9)一种具有式(ii')的化合物:
[0063]
[化学方案12]
[0064][0065]
或其盐。
[0066]
(10)根据(9)的化合物的盐,该盐是甲苯磺酸盐。
[0067]
(11)一种根据(10)的盐的晶体。
[0068]
(12)根据(11)的晶体,其特征在于x射线粉末衍射图,其中至少两个峰的衍射角(2θ)选自由以下各项组成的组:5.9
±
0.2
°
、8.4
±
0.2
°
、11.6
±
0.2
°
、12.7
±
0.2
°
、13.1
±
0.2
°
和15.7
±
0.2
°
。
[0069]
(13)根据(11)的晶体,其特征在于x射线粉末衍射图,该衍射图包括在5.9
±
0.2
°
、8.4
±
0.2
°
、11.6
±
0.2
°
、12.7
±
0.2
°
、13.1
±
0.2
°
和15.7
±
0.2
°
的衍射角(2θ)处的峰。
[0070]
(14)根据(11)的晶体,其特征在于与图4基本相同的粉末x射线衍射谱。
[0071]
(15)一种具有式(iv')的化合物:
[0072]
[化学方案13]
[0073][0074]
或其盐。
[0075]
(16)根据(15)的化合物的盐,该盐是甲磺酸盐。
[0076]
(17)一种根据(16)的盐的晶体。
[0077]
(18)根据(17)的晶体,其特征在于x射线粉末衍射图,其中至少两个峰的衍射角(2θ)选自由以下各项组成的组:7.1
±
0.2
°
、9.3
±
0.2
°
、12.6
±
0.2
°
、14.1
±
0.2
°
、17.7
±
0.2
°
、18.7
±
0.2
°
、19.2
±
0.2
°
、22.2
±
0.2
°
、25.4
±
0.2
°
、27.7
±
0.2
°
、28.5
±
0.2
°
和37.8
±
0.2
°
。
[0078]
(19)根据(17)的晶体,其特征在于x射线粉末衍射图,该衍射图包括在7.1
±
0.2
°
、9.3
±
0.2
°
、12.6
±
0.2
°
、14.1
±
0.2
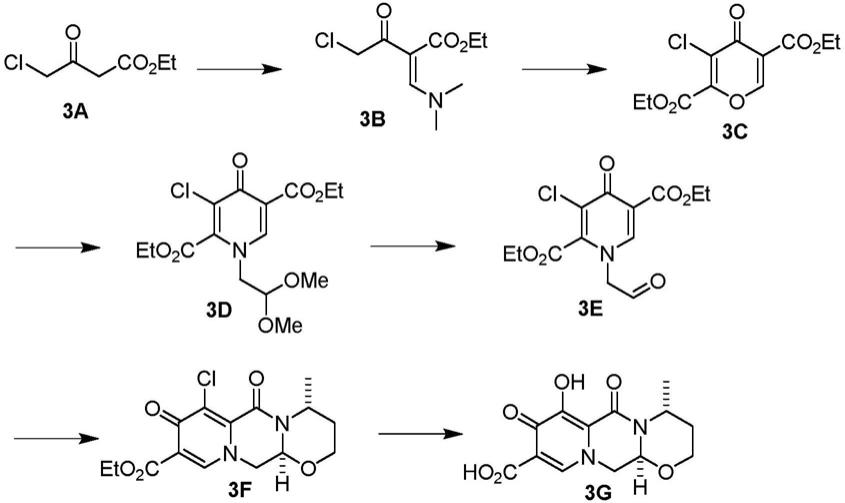
°
、17.7
±
0.2
°
、18.7
±
0.2
°
、19.2
±
0.2
°
、22.2
±
0.2
°
、25.4
±
0.2
°
、27.7
±
0.2
°
、28.5
±
0.2
°
和37.8
±
0.2
°
的衍射角(2θ)处的峰。
[0079]
(20)根据(17)的晶体,在差示扫描量热法中具有219℃
±
2℃的熔点。
[0080]
(21)根据(17)的晶体,其特征在于与图5基本相同的粉末x射线衍射谱。
[0081]
(22)一种具有式(vii)的化合物:
[0082]
[化学方案14]
[0083][0084]
或其盐。
[0085]
(23)一种根据(22)的化合物的一水合物。
[0086]
(24)根据(23)的一水合物,其特征在于x射线粉末衍射图,其中至少两个峰的衍射角(2θ)选自由以下各项组成的组:5.4
±
0.2
°
、7.5
±
0.2
°
、8.4
±
0.2
°
、10.6
±
0.2
°
、11.9
±
0.2
°
、13.5
±
0.2
°
、20.2
±
0.2
°
和22.9
±
0.2
°
。
[0087]
(25)根据(23)的一水合物,其特征在于x射线粉末衍射图,该衍射图包括在5.4
±
0.2
°
、7.5
±
0.2
°
、8.4
±
0.2
°
、10.6
±
0.2
°
、11.9
±
0.2
°
、13.5
±
0.2
°
、20.2
±
0.2
°
和22.9
±
0.2
°
的衍射角(2θ)处的峰。
[0088]
(26)根据(23)的一水合物,其特征在于与图1基本相同的粉末x射线衍射谱。
[0089]
(27)一种具有式(viii)的化合物的溶剂化物:
[0090]
[化学方案15]
[0091][0092]
(28)一种具有式(viii)的化合物的1/2水合物。
[0093]
(29)根据(28)的1/2水合物,其特征在于x射线粉末衍射图,其中至少两个峰的衍射角(2θ)选自由以下各项组成的组:9.5
±
0.2
°
、13.4
±
0.2
°
、18.0
±
0.2
°
、19.3
±
0.2
°
、21.2
±
0.2
°
、22.5
±
0.2
°
、22.8
±
0.2
°
、23.6
±
0.2
°
、27.5
±
0.2
°
和28.1
±
0.2
°
。
[0094]
(30)根据(28)的1/2水合物,其特征在于x射线粉末衍射图,该衍射图包括在9.5
±
0.2
°
、13.4
±
0.2
°
、18.0
±
0.2
°
、19.3
±
0.2
°
、21.2
±
0.2
°
、22.5
±
0.2
°
、22.8
±
0.2
°
、23.6
±
0.2
°
、27.5
±
0.2
°
和28.1
±
0.2
°
的衍射角(2θ)处的峰。
[0095]
(31)根据(28)的1/2水合物,其特征在于与图2基本相同的粉末x射线衍射谱。
[0096]
(32)一种具有式(ix)的化合物:
[0097]
[化学方案16]
[0098][0099]
其盐或其溶剂化物。
[0100]
(33)一种具有式(ix)的化合物的晶体。
[0101]
(34)根据(33)的晶体,其特征在于x射线粉末衍射图,其中至少两个峰的衍射角(2θ)选自由以下各项组成的组:7.1
±
0.2
°
、14.1
±
0.2
°
、15.1
±
0.2
°
、21.0
±
0.2
°
、21.2
±
0.2
°
、22.9
±
0.2
°
和23.4
±
0.2
°
。
[0102]
(35)根据(33)的晶体,其特征在于x射线粉末衍射图,该衍射图包括在7.1
±
0.2
°
、14.1
±
0.2
°
、15.1
±
0.2
°
、21.0
±
0.2
°
、21.2
±
0.2
°
、22.9
±
0.2
°
和23.4
±
0.2
°
的衍射角(2θ)处的峰。
[0103]
(36)根据(33)的晶体,其特征在于与图3基本相同的粉末x射线衍射谱。
[0104]
(37)一种具有式(v)的化合物的晶体:
[0105]
[化学方案17]
[0106][0107]
或其药学上可接受的盐的晶体。
[0108]
(38)根据(37)的晶体,其特征在于x射线粉末衍射图,其中至少两个峰的衍射角(2θ)选自由以下各项组成的组:9.6
±
0.2
°
、10.9
±
0.2
°
、17.8
±
0.2
°
、21.5
±
0.2
°
、22.1
±
0.2
°
、23.5
±
0.2
°
和24.8
±
0.2
°
。
[0109]
(39)根据(37)的化合物的晶体,其特征在于x射线粉末衍射图,该衍射图包括在9.6
±
0.2
°
、10.9
±
0.2
°
、17.8
±
0.2
°
、21.5
±
0.2
°
、22.1
±
0.2
°
、23.5
±
0.2
°
和24.8
±
0.2
°
的衍射角(2θ)处的峰。
[0110]
(40)根据(37)的化合物的晶体,其特征在于与图6基本相同的粉末x
‑
射线衍射谱。
[0111]
根据本发明的方法,具有式(v)或(vi)的多环吡啶酮衍生物可以高光学纯度有效率地制备。
[0112]
[附图简述]
[0113]
图1是化合物3的粉末x射线衍射图。
[0114]
图2是化合物9的粉末x射线衍射图。
[0115]
图3是化合物13的粉末x射线衍射图。
[0116]
图4是化合物20的甲苯磺酸盐的粉末x射线衍射图。
[0117]
图5是化合物21的甲磺酸盐的粉末x射线衍射图。
[0118]
图6是化合物(v)的粉末x射线衍射图。
[0119]
图7是在非禁食条件下将具有式(vi)的化合物(其是具有式(v)的化合物的前药)口服给予至大鼠之后,具有式(v)的化合物在血浆中的浓度的时程。
[0120]
图8是在非禁食条件下将具有式(vi)的化合物(其是具有式(v)的化合物的前药)口服给予至大鼠之后,具有式(vi)的化合物在血浆中的浓度的时程。
[0121]
[发明详述]
[0122]
在下面解释如在此使用的术语的含义。除非另有说明,否则每个术语在单独使用或与其他术语组合使用时具有相同的含义。
[0123]
术语“由......组成”意指仅具有所描述的元素。
[0124]
术语“包含”意指不限于所描述的元素且不排除未描述的元素。
[0125]“卤素”包括氟、氯、溴或碘。氟和氯是优选的,并且氟是特别优选的。
[0126]“烷基”意指c1至c6直链或支链烷基,并且包括c1至c4烷基、c1至c3烷基等。例子包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、己基、异己基等。
[0127]
不同于未取代烷基的保护基团r1的例子包括苄基。
[0128]
未取代烷基r2的例子包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、己基和异己基;并且正丙基、异丁基、己基等是优选的;并且己基是特别优选的。
[0129]“不同于未取代烷基的保护基团”没有限制,只要它是不同于以上“烷基”的保护基团并且它在钠盐和/或镁盐的存在下被移除。例子包括取代烷基以及类似物、优选苄基以及类似物。
[0130]“钠盐”没有限制,只要它能够移除“不同于烷基的保护基团”。例子包括氢氧化钠、氢化钠、异丙基氧化钠、叔戊醇钠、异丙基氯化镁等。叔戊醇钠和异丙基氯化镁是优选的,并且异丙基氯化镁是特别优选的。
[0131]
在以下描述了r1、r2、r3、r4、r5、r6和“钠盐和/或镁盐”的优选实施方案。具有以下实施方案的可能组合的化合物是优选的。
[0132]
r1包括氢或不同于未取代烷基的保护基团。在优选实施方案中,r1是不同于未取代烷基的保护基团,并且苄基是特别优选的。
[0133]
r2包括未取代烷基。在优选实施方案中,r2包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、己基、异己基等,并且正丙基、异丁基、己基等是优选的,并且己基是特别优选的。
[0134]
在优选的实施方案中,“钠盐和/或镁盐”优选是“镁盐”,并且异丙基氯化镁、环己基氯化镁等是更优选的,并且异丙基氯化镁是特别优选的。
[0135]
r3、r4、r5和r6各自独立地为氢或卤素,并且r3、r4、r5和r6中的卤素数量是一个或两个。
[0136]
在一个优选的实施方案中,r3是氢。
[0137]
在一个优选的实施方案中,r4是氢。
[0138]
在一个优选的实施方案中,r5是氟。
[0139]
在一个优选的实施方案中,r6是氟。
[0140]
如在此使用的,术语“r3、r4、r5和r6中的卤素数量是一个或两个”意指r3、r4、r5和r6中的一个或两个是卤素。
[0141]
在本说明书中,使化合物与化合物反应包括使此种化合物的盐或其溶剂化物反应。
[0142]
本发明的化合物的药学上可接受的盐的例子包括与碱金属(例如,锂、钠、钾等)、碱土金属(例如,钙、钡等)、镁、过渡金属(例如,锌、铁等)、氨、有机碱(例如,三甲胺、三乙胺、二环己基胺、乙醇胺、二乙醇胺、三乙醇胺、葡甲胺、乙二胺、吡啶、甲基吡啶、喹啉等)或氨基酸的盐,或者与无机酸(例如,盐酸、硫酸、硝酸、碳酸、氢溴酸、磷酸、氢碘酸等)、或有机酸(例如,甲酸、乙酸、丙酸、三氟乙酸、柠檬酸、乳酸、酒石酸、草酸、马来酸、富马酸、苦杏仁酸、戊二酸、苹果酸、苯甲酸、苯二甲酸、抗坏血酸、苯磺酸、对甲苯磺酸、甲磺酸、乙磺酸等)
的盐,特别是与盐酸、硫酸、磷酸、酒石酸、甲磺酸等的盐。这些盐可以按照常规方法形成。
[0143]
具有式(v)的化合物的药学上可接受的盐的例子包括与碱金属(例如,锂、钠、钾等)、碱土金属(例如,钙、钡等)、镁、过渡金属(例如,锌、铁等)的盐,并且与碱金属(例如,锂、钠、钾等)的盐和与碱土金属(例如,钙、钡等)的盐是优选的。
[0144]
本发明的化合物或其药学上可接受的盐可形成溶剂化物,例如水合物,和/或结晶多晶型物,并且本发明包括此类各种溶剂化物以及结晶多晶型物。“溶剂化物”可以是其中任何数量的溶剂分子(例如,水分子等)与本发明的化合物配位的那些。当允许本发明的化合物或其药学上可接受的盐在大气中静置时,它可以吸收水分,导致吸附水的附着或水合物的形成。此外,本发明的化合物或其药学上可接受的盐可以重结晶以形成晶体多晶型物。
[0145]
下面阐明用于表征本发明的晶体的方法。除非另有说明,否则说明书和权利要求书中的数值是近似值。由于仪器校准、仪器误差、材料纯度、晶体尺寸、样品尺寸和其他因素,数值可能变化。
[0146]
如在此使用的术语“晶体”意指具有有序长程分子结构的材料。晶型的结晶度可通过许多技术测量,包括例如粉末x射线衍射、水分吸附、差示分析、量热分析、溶液比色法、溶解特性。
[0147]
通常,结晶有机化合物由在三维空间中周期性排列的大量原子构成。结构周期性通常表现出通过大多数光谱探针(例如,x射线衍射、红外光谱、拉曼光谱和固态nmr)可明显区分的不同物理特性。
[0148]
其中,x射线粉末衍射(xrpd)被公认是确定固体结晶度的最敏感方法之一。照射到晶体上的x射线被晶格面反射并且相互干涉。然后,只有在满足布拉格定律预测的条件的方向上的衍射线被增强,并且有序衍射线的强度被抵消并且观察不到。在另一方面,在无定形固体的情况下,未观察到长程内的有序衍射线。由于不存在长程有序的重复晶格,无定形固体通常表现出称为晕圈图(halo pattern)的宽xrpd图。
[0149]
本说明书中公开的多环吡啶酮衍生物、其中间体、盐和/或其溶剂化物的晶型优选具有可区分的x射线粉末衍射曲线。例如,具有式(v)化合物的晶型可以优选通过特征衍射峰的存在来区分。如在此使用的特征衍射峰是选自观察到的衍射图中的那些。优选地,特征衍射峰从衍射图中大约二十个峰、更优选大约十个峰并且最优选大约五个峰之中进行选择。
[0150]
一般而言,已知在如下示出的表和图中的各种峰的相对强度可因诸多因素而变化,这些因素例如晶体对x射线束的取向效应、待分析材料的纯度或样品的结晶度。峰位置也可能对于样品高度的变化而发生位移。此外,根据布拉格方程(nλ=2d sinθ),使用不同波长的测量将导致不同的位移。通过使用不同波长获得的此种xprd图在本发明的范围内。
[0151]
本发明的晶型可以通过热分析来表征。
[0152]
dsc(差示扫描量热法)
[0153]
dsc是用于热分析的主要测量方法之一,并且是一种用于测量作为原子/分子聚集体的物质的热特性的方法。差示扫描量热曲线可以通过以下方式获得:通过dsc测量药物活性成分随温度或时间的热容变化,并且将获得的数据对温度或时间绘图。可以从差示扫描量热曲线获得药物活性成分的起始温度、吸热最大值和熔化焓的信息。
[0154]
(本发明的化合物的制备)
[0155]
下面例示了用于制备本发明的化合物的通用方法。此外,萃取、纯化等可以通过有机化学实验中实践的常规方法来进行。
[0156]
本发明的化合物的合成可参考本领域已知的方法进行。
[0157]
作为原料化合物,可以利用可商购化合物、本说明书中描述的化合物、本说明书中引用的参考文献中描述的化合物和其他已知化合物。
[0158]
如果希望本发明化合物的盐,在以盐的形式获得本发明化合物的情况下可以将其按原样进行纯化。在该化合物以游离形式获得的情况下,将其溶解或悬浮在合适的有机溶剂中,并且通过常规方法添加酸或碱以形成盐。
[0159]
此外,本发明化合物及其药学上可接受的盐可以与水或各种溶剂的加合物形式(水合物或溶剂化物)存在。本发明还包括此类加合物。
[0160]
楔形和虚线指示绝对构型。
[0161]
本发明的方法可以例如如下进行。
[0162]
步骤1
[0163]
[化学方案18]
[0164][0165]
其中r1是氢或不同于未取代烷基的保护基团,并且r2是未取代烷基。
[0166]
在此步骤中,在钠盐和/或镁盐的存在下使具有式(i)化合物与具有式r2‑
oh的醇反应,以得到具有式(ii)的化合物。
[0167]
溶剂不受限制,只要其允许以上过程有效率地进行。此种溶剂的例子包括二氯甲烷、甲苯、四氢呋喃等,它们可以单独使用或组合使用。该反应可以在单一混合溶剂中、或者在没有溶剂的情况下进行。优选的溶剂是四氢呋喃。
[0168]
钠盐和/或镁盐的例子包括氢氧化钠、氢化钠、异丙醇钠、叔戊醇钠、异丙基氯化镁、环己基氯化镁等。优选的是异丙基氯化镁。该盐可以相对于化合物(i)0.1至5摩尔当量、优选0.3至0.5摩尔当量的量使用。
[0169]
反应温度没有限制,但反应通常可在约0℃至100℃、优选在0℃至室温下进行。
[0170]
反应时间没有限制,但反应通常可进行0.5小时至24小时、优选1至10小时。
[0171]
步骤2
[0172]
[化学方案19]
[0173][0174]
其中r3、r4、r5和r6各自独立地是氢或卤素,条件是r3、r4、r5和r6中的一个或两个是卤素。其他符号是如上所定义的。
[0175]
在此步骤中,具有式(ii')的化合物在缩合剂存在下与具有式(iii)的化合物反应,以获得具有式(iv)化合物。
[0176]
溶剂不受限制,只要其允许以上步骤有效率地进行。溶剂的例子包括乙酸乙酯、环己烷、乙酸异丙酯、乙酸丙酯、甲苯、1,4
‑
二噁烷、dma、dmf、甲苯、庚烷、环戊基甲基醚等,它们可以单独使用或组合使用。该反应可以在单一混合溶剂中、或者在没有溶剂的情况下进行。优选的溶剂是乙酸乙酯和环己烷的混合溶剂。
[0177]
缩合剂的实例包括丙基膦酸酐、甲磺酸、三氟乙酸、对甲苯磺酸一水合物、10
‑
樟脑磺酸、浓硫酸,二氯乙酸、四甲基硫酸氢铵等,并且它们可以单独使用或组合使用,优选丙基膦酸酐和甲磺酸的混合物。缩合剂可以相对于化合物(ii')1至5摩尔当量、优选1至3摩尔当量的量使用。
[0178]
反应温度没有特别限制,但反应通常可在约0℃至100℃、优选在0℃至室温下进行。
[0179]
反应时间没有限制,但反应通常可进行0.5小时至24小时、优选1至10小时。
[0180]
步骤3
[0181]
[化学方案20]
[0182][0183]
其中变量是如上所定义的。
[0184]
在此步骤中,使具有式(iv)化合物与金属盐反应,以获得具有式(iv”)的化合物。
[0185]
溶剂不受限制,只要其允许以上过程有效率地进行。此种溶剂的例子包括n
‑
甲基吡咯烷酮、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺等,它们可以单独使用或组合使用。n
‑
甲基吡咯烷酮是优选的。
[0186]
金属盐的例子包括氯化锂和溴化锂等,并且氯化锂是优选的。金属盐可以相对于
化合物(iv)1至20摩尔当量、优选5至10摩尔当量的量使用。
[0187]
反应温度没有特别限制,但反应通常可在约0℃至100℃、优选在室温至100℃下进行。
[0188]
反应时间没有限制,但反应通常可进行0.5小时至48小时、优选12至24小时。
[0189]
步骤4
[0190]
[化学方案21]
[0191][0192]
其中p
r
是羟基的保护基团,如酯基或醚基,并且其他变量是如上所定义的。
[0193]
在此步骤中,化合物(v”')可根据用于将化合物(iv”)的羟基转化为酯基或醚基的常规方法获得。此种方法的例子可在protective groups in organic synthesis,theodora w green(john wiley&sons),prog.med 5:2157
‑
2161(1985)以及大英图书馆提供的
‑“
the world's knowledge”中找到。
[0194]
如在此使用的“非对映异构体比率”是指下面所示的两种立体异构体之间的hplc面积百分比的比率。
[0195]
[化学方案22]
[0196][0197]
具有式(v)的化合物可用于由流感病毒诱发的症状和/或疾病。它可用于治疗、预防和/或症状缓解:例如,涉及发烧、寒颤、头痛、肌肉疼痛和全身无足轻重感觉(feeling of generalized worthlessness)的感冒症状,气道炎症,如咽喉痛、鼻涕、鼻塞、咳嗽、痰,胃肠道症状,如胃痛、呕吐、腹泻、此外,涉及继发感染的伴发性疾病,如急性脑病、肺炎。
[0198]
由式(vi)表示的化合物可以是优异的药物,因为其具有高口服吸收性、良好的生物利用度、良好的清除率、高肺迁移(pulmonary migration)等优点。
[0199]
由式(v)表示的化合物可以是副作用降低的药物,因为它对作为病毒特异性酶的帽依赖性核酸内切酶具有高抑制活性,并且因此具有高特异性作用。
[0200]
此外,具有式(v)的化合物和/或具有式(vi)的化合物在代谢稳定性、高溶解度、高
口服吸收性、良好生物利用度、良好清除率、高肺转移性(lung transitivity)、长半衰期、对非蛋白质的高结合率、低herg通道抑制、低cyp抑制、cpe(细胞病变效应)抑制作用方面是优异的,和/或它还具有在光毒性测试、埃姆斯(ames)测试、遗传毒性测试中为阴性或没有毒性如肝损伤的优点。因此,具有式(v)的化合物和/或具有式(vi)的化合物可以是优异的药剂。
[0201]
具有式(v)的化合物和/或具有式(vi)的化合物可以通过口服或肠胃外途径给予。对于口服给予,具有式(v)的化合物和/或具有式(vi)的化合物可以任何形式的通常配制品使用,例如,呈固体形式的配制品,例如片剂、粉末、颗粒、胶囊;呈液体形式的配制品,如水性配制品;油性悬浮液;糖浆或酏剂。对于肠胃外给予,具有式(v)的化合物和/或具有式(vi)的化合物可以水性或油性悬浮注射剂或滴鼻剂的形式使用。在此种配制品的制备中,可任选使用常规赋形剂、粘合剂、润滑剂、水性溶剂、油性溶剂、乳化剂、悬浮剂、防腐剂、稳定剂等。包含具有式(v)的化合物和/或具有式(vi)的化合物的药物组合物可通过将治疗有效量的具有式(v)的化合物和/或具有式(vi)的化合物与药学上可接受的载体或稀释剂组合(例如,共混)来制备。
[0202]
对于口服给予,具有式(v)的化合物和/或具有式(vi)的化合物的每日剂量对于成人可为约0.05
‑
3000mg、优选约0.1
‑
1000mg/天,而此剂量取决其给予途径;患者的年龄、体重、病症;和患者疾病而变化。必要时可以将该剂量分开给予。在肠胃外给予的情况下,成人的每日剂量可以为在约0.01
‑
1000mg、优选约0.05
‑
500mg之间。
[实施例]
[0203]
参照实施例、参考实施例、中间体的制备实施例和测试实施例更详细地解释本发明,但本发明不限于这些实施例。
[0204]
参考实施例和实施例中的nmr分析在400mhz下使用dmso
‑
d6、cdcl3进行。
[0205]
粉末x射线衍射图
[0206]
在以下条件下根据日本药典中的通用测试中的粉末x射线衍射分析方法进行每个实施例中获得的晶体的粉末x射线衍射分析。
[0207]
(装置)
[0208]
minflex600 rint
‑
ttriii(日本理学株式会社(rigaku))
[0209]
(方法)
[0210]
检测器:高速一维检测器(d/tecultra 2)和可变刀刃(variable knife edge)
[0211]
测量方法:反射法
[0212]
光源类型:cu灯泡
[0213]
工作波长:cukα射线
[0214]
管电流:10ma或15ma
[0215]
管电压:30kv,或40kv
[0216]
样品板:铝或玻璃
[0217]
x射线入射角(θ):3
‑
40
°
,采样宽度:0.01
°
,或
[0218]
x射线入射角(θ):4
‑
40
°
,采样宽度:0.02
°
[0219]
通常,由于粉末x射线衍射中的衍射角(2θ)可能涉及
±
0.2
°
内的误差,因此衍射角
的值包括在约
±
0.2
°
范围内的值。因此,本发明不仅包括其中粉末x射线衍射中的峰的衍射角完全匹配的晶体,而且还包括其中峰的衍射角与约
±
0.2
°
的误差一致的晶体。
[0220]
(通过卡尔费休法测量含水量)
[0221]
根据日本药典中的水测定(库仑滴定)的通用测试来测定含水量。aquamicron
tm ax(三菱化学株式会社(mitsubishi chemical corporation))用作阳极电解质溶液,并且aquamicron
tm cxu用作阴极电解质溶液。
[0222]
通常,通过卡尔费休法的含水量测量可能涉及在
±
0.3%范围内的误差。因此,测量的含水量的具体值应包括
±
0.3%范围内的任何值。
[0223]
tg/dta测量
[0224]
进行每个实施例中获得的晶体的tg/dta测量。将样品在铝盘中称重并在开放系统中测量。测量条件如下所示。
[0225]
设备:tg/dta 7200(日立高科技公司(hitachi high
‑
tech science))
[0226]
测量温度范围:30℃
‑
250℃
[0227]
加热速率:10℃/min
[0228]
通常,tg/dta测量可能涉及
±
2℃范围内的误差。因此,测量的特定值应包括
±
2℃范围内的任何值。
[0229]
动态蒸气吸附(dvs)
[0230]
进行每个实施例中获得的晶体的动态蒸气吸附分析。将样品在样品盘中称重并在如下条件下测量。
[0231]
设备:dvs advantage(表面测量系统有限公司(surface measurement systems ltd.))
[0232]
测量点:从0%rh以5%步进至95%rh,然后95%rh以5%步进至0%rh
[0233]
温度:25℃或60℃
[0234]
差示扫描量热法(dsc)的测量
[0235]
进行每个实施例中获得的晶体的dsc测量。将样品在具有气密密封的不锈钢盘中称重并在以下条件下测量。
[0236]
设备:mettler toledo dsc 822e
[0237]
测量温度范围:30℃
‑
300℃
[0238]
加热速率:10℃/min
[0239]
气氛:n2 40ml/min
[0240]
通常,差示扫描量热法(dsc)可能涉及在
±
2℃范围内的误差。因此,如通过差示扫描量热法(dsc)测量的特定值应包括在
±
2℃范围内的任何值。
[0241]
实施例中每个术语的含义如下。
[0242]
dma:n,n
‑
二甲基乙酰胺
[0243]
thf:四氢呋喃
[0244]
t3p:丙基膦酸酐(环状三聚体)
[0245]
实施例1:化合物3的制备
[0246]
[化学方案23]
[0247][0248]
步骤1:化合物3
[0249]
将dma(300ml)添加到化合物1(100.00g,406mmol)中并且搅拌混合物。添加碳酸氢钠(44.41g,529mmol)、硫酸二甲酯(58.91g,467mmol)和dma(100ml)并且在25℃下搅拌7小时。将合成盐酸(16.90g)和水(500g)添加到反应混合物中,并且将该混合物用乙酸乙酯(1000ml和550ml)萃取两次。用5%盐水(300g)和水(300g)洗涤有机层。在减压下将合并的有机层浓缩至约500g。将乙酸乙酯(350ml)添加到浓缩物中,并且在减压下将所得溶液浓缩至约500g。将dma(300ml)添加到浓缩物中,并且在减压下将所得溶液浓缩至约400g。将对甲苯磺酸吡啶鎓(265.42g)和dma(100ml)添加到浓缩物中,并且将反应混合物加热至60℃。在6小时内将肼基甲酸叔丁酯(tert
‑
butyl carbazinate)(69.80g,528mmol)的dma(100ml)溶液缓慢添加到反应混合物中。将反应混合物在60℃下搅拌3小时并冷却至25℃。将乙醇(100ml)和水(290ml)添加到反应混合物中,并且将反应混合物温热至30℃。将乙醇(100ml)和水(520ml)的混合物缓慢添加到反应混合物中。将反应混合物冷却至0℃,并且然后在0℃下搅拌1.5小时。通过过滤收集所得的淡黄白色沉淀物。将所得固体用乙醇(480ml)和水(720ml)的混合物洗涤并干燥,以得到呈浅黄白色固体的化合物3的一水合物(122.70g,产率77%)。
[0250]1h
‑
nmr(400mhz,cdcl3)δ:1.45(s,9h),3.77(s,3h),5.26(s,2h),6.39(d,j=7.6hz,1h),7.27
‑
7.47(m,6h),7.64
‑
8.23(br s,1h)
[0251]
粉末x射线衍射2θ(
°
):5.4、7.5、8.4、10.6、11.9、13.5、20.2、22.9
[0252]
化合物3的粉末x射线衍射图示于图1中。
[0253]
通过卡尔费休法的含水量:4.5%
[0254]
实施例2:化合物9的制备
[0255]
[化学方案24]
[0256]
[0257]
步骤1:化合物6
[0258]
将化合物5(28.29g,167.4mmol)和dma(65ml)添加到化合物4(20.00g,104.6mmol)中,并且搅拌混合物。在将该混合物温热至40℃后,缓慢添加叔丁醇钠(15.09g,157.0mmol)。将反应混合物在40℃下搅拌3小时并且然后冷却至20℃。将乙酸(3.14g)和10%氯化钠水溶液(64g)添加到反应混合物中,并且将混合物用乙酸乙酯(60ml)萃取两次。将水(144ml)添加到合并的有机层中,并且将混合物冷却至0℃。通过过滤收集所得的淡黄白色沉淀物。将所得固体用甲醇(5.4g)和水(48.6g)的混合物洗涤,并且干燥,以得到呈淡黄白色固体的化合物6(20.44g,产率78%)。
[0259]1h
‑
nmr(cdcl3)δ:3.34(s,6h),3.53(d,j=5.2hz,2h),3.76(t,j=5.6hz,2h),3.90(t,j=5.6hz,2h),4.43(t,j=5.2hz,1h),7.70
‑
7.73(m,2h),7.84
‑
7.87(m,2h)
[0260]
步骤2化合物8
[0261]
将乙醇(20ml)和水(20ml)添加到化合物6(20.02g,71.68mmol)中,并且搅拌混合物。将该混合物温热至60℃。向该混合物中添加60%一水合肼水溶液(8.99g,107.7mmol)并且在60℃下搅拌4小时。在添加水(40ml),然后冷却至30℃之后,将17%氢氧化钾水溶液(92.12g)添加到反应混合物中。将反应混合物用二氯甲烷(120、78、78、78ml)萃取四次。将合并的有机层用水(20ml)洗涤,并且在减压下浓缩至约160g。将thf(100ml)添加到浓缩物中,并且在减压下将混合物浓缩至约40g。将thf(100ml)添加到浓缩物中,并且在减压下将混合物浓缩至约40g。将thf(20ml)添加到浓缩物中,并且在减压下将混合物浓缩至约15g,以获得15g化合物7的thf溶液。
[0262]
将以上化合物7(14.71g)、thf(7g)和1,8
‑
二氮杂双环[5.4.0]
‑7‑
十一烯(379.0mg)的thf溶液添加到化合物3(10.00g,25.5mmol)中,并且搅拌混合物。将反应混合物加热至60℃,并且然后在60℃下搅拌24小时。在将反应混合物冷却至25℃之后,添加水(28g)和乙酸(3.72g)。将反应混合物用乙酸乙酯(50ml、30ml)萃取两次,并且将有机层用5%碳酸氢钠水溶液(30g)和水(28g)洗涤。在减压下将有机层浓缩至约36g。将乙酸乙酯添加到反应混合物中,并且在减压下将所得混合物浓缩至约36g。将庚烷(65ml)添加到浓缩物中,并且将混合物冷却至5℃。在5℃下搅拌1小时之后,通过过滤收集所得的淡黄白色沉淀物。将所得固体用庚烷(32ml)和乙酸乙酯(14ml)的混合物洗涤并干燥,以获得呈淡黄白色固体的化合物8(10.10g,产率81%)。
[0263]1h
‑
nmr(cdcl3)δ:1.44(s,9h),3.32
‑
3.48(m,12h),4.41(t,j=5.2hz,1h),5.29(s,2h),6.38(d,j=7.6hz,1h),7.11
‑
7.50(m,7h),8.46(s,1h).
[0264]
步骤3:化合物9
[0265]
将乙腈(170ml)和水(30ml)添加到化合物8(19.99g,40.7mmol)中,并且搅拌混合物。将反应混合物加热至60℃,并且缓慢添加甲磺酸(11.70g,121.7mmol)。将反应混合物在60℃下搅拌6小时并且然后冷却至25℃。将30%氢氧化钠水溶液(15.91g)添加到反应混合物中,并且在减压下将所得混合物浓缩至约100g。将水(50ml)添加到浓缩物中,并且在减压下将所得混合物浓缩至约100g。在将浓缩物在25℃下搅拌30分钟之后,通过过滤收集所得的黄色沉淀物。将所获得的固体用水(40ml)洗涤并干燥,以获得呈黄色晶体的化合物9的0.5水合物(10.43g,产率76%)。
[0266]1h nmr(400mhz,dmso
‑
d6),δ:2.95(ddd,j=13.7,12.3,4.3hz,1h),3.13(dd,j=
11.2,10.0hz,1h),3.44(td,j=11.9,3.1hz,1h),3.96
‑
4.08(m,2h),4.14(dd,j=13.9,2.4hz,1h),4.80(ddd,j=12.6,9.9,4.5hz,1h),5.08(s,2h),6.22(d,j=7.6hz,1h),7.24
‑
7.41(m,4h),7.52
‑
7.60(m,2h),7.69(d,j=7.6hz,1h)
[0267]
粉末x射线衍射2θ(
°
):9.5、13.4、18.0、19.3、21.2、22.5、22.8、23.6、27.5、28.1
[0268]
化合物9的粉末x射线衍射图示于图2中。
[0269]
通过卡尔费休法的含水量:2.8%
[0270]
实施例3化合物13的方法
[0271]
[化学方案25]
[0272][0273]
步骤1:化合物11和12
[0274]
将乙酸乙酯(87ml)和50(w/w)%t3p乙酸乙酯溶液(145.80g,229.1mmol)添加到化合物9的0.5水合物(30.00g,89.2mmol)中,并且搅拌混合物。将反应混合物加热至60℃,添加三乙胺(18.55g,183.3mmol),并且然后缓慢添加(r)
‑
( )
‑
四氢呋喃
‑2‑
甲酸(12.24g,105.4mmol)。将反应混合物在60℃下搅拌4.5小时并然后冷却至0℃,并且通过过滤收集所得的淡黄色沉淀物。将所获得的固体用乙酸乙酯(120ml)洗涤,以获得呈淡黄色固体的化合物11(18.34g,未干燥的)。而且,将滤液和洗涤溶液合并,以获得化合物12的乙酸乙酯溶液(358.60g)。
[0275]
步骤2:化合物13和9
[0276]
将乙酸乙酯(120ml)和1,8
‑
二氮杂双环[5.4.0]
‑7‑
十一烯(530mg,3.5mmol)添加到化合物11(15.28g)中,并且搅拌混合物。将反应混合物加热至30℃,并且缓慢添加甲醇(1.67g)和乙酸乙酯(43ml)的混合物。将反应混合物在室温下搅拌1小时,并且通过过滤收集所得的白色沉淀物。将所获得的晶体用乙酸乙酯(60ml)洗涤并干燥,以获得化合物13的白色结晶(11.06g,产率45%)。
[0277]1h
‑
nmr(cdcl3)δ:2.84
‑
2.92(m,2h),3.45(td,j=3.2hz,12.0hz,1h),3.82(dd,j=4.0hz,11.2hz,1h),3.92(dd,j=4.4hz,11.6hz,1h),4.13(dd,j=2.8hz,13.6hz,1h),4.47
‑
4.54(m,1h),4.96(d,j=9.6hz,1h),5.27(d,j=10.0hz,1h),5.76(d,j=13.2hz,1h),6.19(d,j=7.6hz,1h),7.22(d,j=8.0hz,1h),7.30
‑
7.38(m,3h),7.59(dd,j=1.6hz,
8.0hz,2h).
[0278]
粉末x射线衍射2θ(
°
):7.1、14.1、15.1、21.0、21.2、22.9、23.4
[0279]
化合物13的粉末x射线衍射图示于图3中。
[0280]
在减压下将化合物12的乙酸乙酯(334.69g)溶液浓缩至约170g。将浓缩溶液在25℃下搅拌。将乙腈(224ml)、水(56ml)和24%氢氧化钠水溶液(150g)缓慢添加到混合物中,并且然后分离成有机层和水层。将水(14ml)添加到水层中并且用乙腈(168ml)萃取两次。在减压下将合并的有机层浓缩至约250g。将浓缩物加热至60℃,并且添加1,8
‑
二氮杂双环[5.4.0]
‑7‑
十一烯(19.01g,124.9mmol)。将反应混合物在60℃下搅拌3.5小时并且然后冷却至40℃。将5.8%盐酸水溶液(50.40g)添加到反应混合物中,并且将所得的混合物冷却至25℃,以获得溶液(314.96g)。在减压下将该溶液的一部分(158.86g)浓缩至约85g。将浓缩物在20℃下搅拌2小时,并且添加水(28ml)。在减压下将反应混合物浓缩至约100g。在将浓缩物在20℃下搅拌1小时之后,通过过滤收集沉淀的淡黄白色晶体。将所获得的晶体用水(42ml)洗涤并干燥,以获得呈淡黄白色晶体的化合物9(5.93g,产率42%)。
[0281]
实施例4:化合物19
[0282]
[化学方案26]
[0283][0284]
步骤1:化合物15
[0285]
将二异丙胺(7.69g,76.0mmol)添加到thf(25ml)中,并且将混合物搅拌并冷却至
‑
40℃。在添加1.6mol/l正丁基锂(43.5ml,69.6mmol)之后,将所得混合物在0℃下搅拌1小时。将混合物冷却至
‑
40℃,并且缓慢添加3,4
‑
二氟苯甲酸(5.00g,31.6mmol)的thf(25ml)溶液。将反应混合物在
‑
40℃下搅拌1小时,并且缓慢添加n,n
‑
二甲基甲酰胺(5.74g,78.5mmol)。向反应混合物中添加6mol/l盐酸水溶液(34.25ml),并且将混合物温热至25℃并且分离成有机层和水层。用乙酸乙酯(15ml)萃取水层。将合并的有机层用水(5ml)洗涤。在减压下浓缩之后,将甲苯添加到残余物中,以获得化合物15的甲苯溶液。
[0286]
步骤2:化合物16
[0287]
将甲苯(17.8ml)、苯硫酚(3.90g,35.4mmol)和d
‑
樟脑磺酸(1.16g,5.0mmol)添加到以上化合物15的溶液中。将混合物搅拌并加热至60℃。将反应混合物在60℃下搅拌4小时并且然后冷却至5℃。将2mol/l氢氧化钠溶液(10ml)添加到反应混合物中,并且将所得的混合物温热至25℃。用甲苯(10ml)萃取反应混合物,并且用2mol/l氢氧化钠(5ml)和水(10ml)洗涤有机层。在减压下浓缩有机层之后,添加甲苯以获得化合物16的甲苯溶液。
[0288]
步骤3:化合物17
[0289]
将氯化铝(5.52g,41.4mmol)和甲苯(25ml)的混合物搅拌并冷却至0℃。将1,1,3,3
‑
四甲基二硅氧烷(5.56g,41.4mmol)的甲苯(10ml)溶液滴加到反应混合物中,并且将混合物温热至25℃。将以上化合物16的甲苯溶液缓慢添加到反应混合物中,并且将混合物在25℃下搅拌2.5小时。在添加15%硫酸水溶液(35ml)后,将混合物搅拌并且然后分离成有机层和水层。将有机层用水(20ml)洗涤两次。在减压下将溶液浓缩至约16g。将庚烷(40ml)缓慢添加到浓缩物中并冷却至0℃。通过过滤收集所得的白色沉淀物。将所获得的固体用庚烷(20ml)洗涤并且然后干燥,以获得呈白色固体的化合物17(7.20g,产率81.3%)。
[0290]1h
‑
nmr(cdcl3)δ:4.61(d,j=1.6hz,2h),7.09
‑
7.15(m,1h),7.23
‑
7.27(m,3h),7.34
‑
7.37(m,2h),7.84
‑
7.88(m,1h)
[0291]
步骤4:化合物18
[0292]
将多磷酸(425.0g)搅拌并且加热至80℃。添加化合物17(85.0g),并且将混合物温热至120℃并在120℃下搅拌3小时。将反应混合物冷却至80℃,并且缓慢添加水(200ml)。将反应混合物冷却至30℃,并且添加水(850ml)。用乙酸乙酯(850ml)萃取混合物。用水(425ml)和10%碳酸氢钠水溶液(255ml)洗涤有机层。将溶剂在减压下蒸发,并且将庚烷(340ml)添加到所获得的残余物中。将溶剂在减压下蒸发,并且将庚烷(85ml)添加到所获得的残余物中。在将反应混合物在30℃下搅拌30分钟之后,通过过滤收集所得棕色沉淀物。将所获得的固体用庚烷(42ml)洗涤并且然后干燥,以获得呈棕色固体的化合物18(72.0g,产率91%)。
[0293]1h
‑
nmr(cdcl3)δ:4.14(d,j=1.0hz,2h),7.09
‑
7.18(m,1h),7.27
‑
7.33(m,1h),7.34
‑
7.45(m,3h),8.19(dd,j=8.5hz,1.4hz,1h)
[0294]
步骤5:化合物19
[0295]
将硼氢化钠(234.0mg,6.2mmol)悬浮于0.5%氢氧化钠水溶液(1.8ml)中以制备硼氢化钠悬浮液。将2
‑
丙醇(20ml)和水(2.25ml)添加到化合物18(4.5g,17.2mmol)中。将混合物搅拌并且加热至40℃。将以上硼氢化钠悬浮液缓慢添加混合物中。将反应混合物在40℃下搅拌1.5小时并冷却至25℃。将水(32ml)添加到反应混合物中,然后添加水(6.7ml)和62%硫酸水溶液(460mg)的混合溶液。将反应混合物冷却至5℃,并且通过过滤收集所得的棕色沉淀物。将固体用水(18ml)洗涤并且然后干燥,以获得呈棕色固体的化合物19(4.4g,产率97%)。
[0296]1h
‑
nmr(cdcl3)δ:2.67(d,j=3.8hz,1h),4.20(dd,j=14.4,1.4hz,2h),4.68(dd,j=14.5,1.3hz,2h),7.02(dt,j=9.7,8.3hz,1h),7.12
‑
7.21(m,4h),7.44
‑
7.49(m,1h)
[0297]
实施例5:化合物(v)和(vi)
[0298]
[化学方案27]
[0299][0300]
步骤1
‑
1:化合物20
[0301]
将1
‑
己醇(22.5g,220mmol)和thf(24.6g)合并,并且将混合物的温度调节至20℃。将异丙基氯化镁的thf溶液(2mol/l,7.2g,14.7mmol)添加到混合物中,以制备六氧化镁溶液。
[0302]
在搅拌下将1
‑
己醇(22.5g,220mmol)添加到化合物13(12.0g,36.7mmol)中,并且将混合物的温度调节至20℃。将以上六氧化镁溶液添加到所得的化合物13的浆料中。将反应混合物在20℃下搅拌4小时,并且然后添加柠檬酸水溶液(3.1g柠檬酸一水合物和36g水)。用thf(10.7g)萃取混合物,并且用水(24g)洗涤有机层。在减压下将有机层浓缩至约55g。将对甲苯磺酸的thf溶液(7.0g对甲苯磺酸一水合物和42.8g thf)添加到所得浓缩物中。在减压下将混合物浓缩至约61g。将thf(42.7g)添加到浓缩物中,并且在减压下将所得混合物浓缩至约61g。在将混合物加热至50℃之后,添加甲基叔丁基醚(133.0g)。将所得混合物冷却至10℃,并且在10℃下搅拌1.5小时。通过过滤收集所得的白色沉淀物。将所获得的固体用甲基叔丁基醚(40.0g)和乙酸乙酯(16.0g)的混合物洗涤并干燥,以获得呈白色晶体的化合物20的甲苯磺酸盐(15.8g,产率87.2%)。
[0303]1h
‑
nmr(cdcl3)δ:0.88(t,j=7.2hz,3h),1.25
‑
1.34(m,4h),1.34
‑
1.43(m,2h),1.76
‑
1.85(m,2h),2.34(s,3h),3.04(ddd,j=13.6,11.7,4.3hz,3h),3.36(dd,j=11.6,10.0hz,3h),3.43(ddd,j=13.6,12.0,4.4hz,3h),4.00(dd,j=11.7,4.3hz,1h),4.06
‑
4.18(m,4h),4.80(br,s,1h),7.16(d,j=7.8hz,1h),7.62(d,j=7.8hz,1h),7.62(d,j=7.1hz,1h),8.17(d,j=7.1hz,1h),8.40(br,s,1h)。
[0304]
粉末x射线衍射2θ(
°
):5.9、8.4、11.6、12.7、13.1、15.7
[0305]
化合物20的粉末x射线衍射图示于图4中。
[0306]
步骤1
‑
2:化合物20
[0307]
使用环己基氯化镁的thf溶液(16.2wt%,0.4当量)代替异丙基氯化镁的thf溶液(0.4当量),如步骤1
‑
1中所述进行反应,并且通过hplc分析反应混合物以确定化合物20的形成速率。
[0308]
化合物20的hplc面积百分比:90.9%(rt=11.0min)
[0309]
其他程序与如步骤1
‑
1中描述的相同。
[0310]
(测量条件)
[0311]
(1)柱:x select
tm csh c18(3.5μm内径4.6
×
100mm)(沃特世公司(waters))
[0312]
流速:1.0ml/min;紫外检测波长:254nm;
[0313]
流动相:
[0314]
[a]0.1%甲酸水溶液,[b]乙腈
[0315]
梯度程序:([b]的浓度)15%
‑
15%5min;15%
‑
60%10min;60%
‑
85%2min;85%
‑
85%3min。
[0316]
步骤1
‑
3:化合物20
[0317]
将1
‑
己醇(27.5g,270mmol)添加到化合物13(4.91g,15.0mmol)中,并且搅拌混合物。将混合物的温度调节至0℃。向所得浆料中添加叔戊醇钠的thf溶液(1.4mol/l,45.0mmol)。在0℃下搅拌2.5小时之后,通过hplc分析反应混合物以确定化合物20的形成速率。
[0318]
化合物20的hplc面积百分比:93.3%(rt=9.5min)
[0319]
(测量条件)
[0320]
(1)柱:chiralpak
tm ib(5.0μm内径4.6
×
250mm)(大赛璐公司(daicel))
[0321]
流速:1.0ml/min;紫外检测波长:254nm;
[0322]
流动相:[a]0.1%甲酸,[b]乙腈
[0323]
梯度程序:用35%溶剂[b]保持5min;在6min内用35%至85%溶剂[b]的线性梯度;并且用85%溶剂[b]维持2min。
[0324]
如上所示,发现当使用镁盐或钠盐时,反应以良好的产率进行。以高产率获得所希望的产物,特别是当使用异丙基氯化镁时。
[0325]
步骤2:化合物21的甲磺酸盐
[0326]
将化合物19(8.0g,30.3mmol)、乙酸乙酯(48.7g)和环己烷(14.1g)添加到化合物20(12.0g,24.3mmol)中,并且将混合物在25℃下搅拌。添加50(w/w)%t3p乙酸乙酯溶液(20.91g,32.9mmol),然后添加甲磺酸(3.5g,36.4mmol)。将混合物加热至60℃并搅拌24小时。在冷却至25℃之后,添加thf(32.0g)和水(24.0g),并且然后缓慢添加24%氢氧化钠水溶液(30.8g)。在沉降之后,将混合物分离成有机层和水层。将有机层用7%氯化钠水溶液(60.0g)洗涤两次。将甲磺酸(2.80g,29.1mmol)的环己烷(9.3g)和乙酸乙酯(32.1g)溶液添加到合并的有机层中。将混合物在25℃下搅拌2小时,并且通过过滤收集所得白色沉淀物。
将所得固体用乙酸乙酯(43.3g)洗涤并且然后干燥,以获得呈白色晶体的化合物21的甲磺酸盐(13.65g,产率84.6%)。
[0327]1h
‑
nmr(dmso
‑
d6)δ:0.90(3h,t,j=6.0hz),1.29
‑
1.36(4h,m),1.39
‑
1.49(2h,m),1.67
‑
1.79(2h,m),2.38(3h,s),2.94(1h,br s),3.30(1h,td,j=11.6,2.4hz),3.51(1h,t,j=10.4hz),3.66(1h,dd,j=11.2,2.8hz),3.92
‑
4.01(2h,m),4.07(1h,d,j=14.3hz),4.20(1h,s),4.42
‑
4.52(1h,m),5.43(1h,dd,j=14.4,2.1hz),5.79
‑
5.83(2h,m),6.81(1h,td,j=7.6,1.2hz),6.96(1h,dd,j=7.8,1.0hz),7.09(1h,j=8.0,1.6hz),7.12
‑
7.18(1h,m),7.32(1h,d,j=7.7hz),7.37
‑
7.49(2h,m)
[0328]
粉末x射线衍射2θ(
°
):7.1、9.3、12.6、14.1、17.7、18.7、19.2、22.2、25.4、27.7、28.5、37.8
[0329]
化合物21的粉末x射线衍射图示于图5中。
[0330]
dsc:开始216℃,峰219℃。
[0331]
步骤3:化合物(v)
[0332]
将氯化锂(8.6g,203.3mmol)添加到n
‑
甲基吡咯烷酮(52.4g)和化合物21(15.0g,22.6mmol)的混合物中,并且将所得混合物加热至75℃。将混合物在75℃下搅拌20小时,并且然后冷却至40℃。将乙腈(20.0g)添加到反应混合物中,然后添加水(11.6g)。在将混合物冷却至30℃并搅拌30分钟之后,缓慢添加水(142.5g)。在30℃下搅拌1.5小时后,通过过滤收集所得白色沉淀物。将所得固体用2
‑
丙醇(60.1g)洗涤并且然后干燥,以获得呈白色晶体的化合物(v)(9.91g,产率90.7%)。
[0333]1h
‑
nmr(cdcl3)δ:3.00(td,j=11.8,3.2hz,1h),3.46(td,j=12.0,2.8hz,1h),3.59(t,j=10.0hz,1h),3.82(dd,j=12.2,3.0hz,1h),3.96(dd,j=11.0,3.0hz,1h),4.07(d,j=13.6hz,1h),4.58(dd,j=10.0,2.8hz,1h),4.67(dd,j=13.6,2.0hz,1h),5.26
‑
5.30(m,2h),5.75(d,j=8.0hz,1h),6.69(d,j=7.6hz,1h),6.83
‑
6.87(m,1h),6.99
‑
7.04(m,2h),7.07
‑
7.15(m,3h).
[0334]
粉末x射线衍射2θ(
°
):9.6、10.9、17.8、21.5、22.1、23.5、24.8
[0335]
具有化合物(v)的粉末x射线衍射图示于图6中。
[0336]
步骤4:化合物(vi)
[0337]
将氯甲基碳酸甲酯(0.483g,3.10mmol)、碳酸钾(0.572g,4.14mmol)和碘化钾(0.343g,2.07mmol)与化合物(v)(1.00g,2.07mmol)于dma中的悬浮液混合(5ml)。将混合物加热至50℃并搅拌6小时。将dma(1ml)添加到反应混合物中,并且将所得的混合物搅拌6小时。将反应混合物冷却至室温,并且添加dma(6ml)。将混合物在50℃下搅拌5分钟,并且然后过滤。在冰冷却下将1mol/l盐酸(10ml)和水(4ml)滴加到所得滤液中,并且然后将混合物搅拌1小时。通过过滤收集沉淀的固体,并且在60℃下在减压下干燥3小时,以获得化合物(vi)(1.10g,1.93mmol,产率93%)。
[0338]1h
‑
nmr(dmso
‑
d6)δ:2.91
‑
2.98(1h,m),3.24
‑
3.31(1h,m),3.44(1h,t,j=10.4hz),3.69(1h,dd,j=11.5,2.8hz),3.73(3h,s),4.00(1h,dd,j=10.8,2.9hz),4.06(1h,d,j=14.3hz),4.40(1h,d,j=11.8hz),4.45(1h,dd,j=9.9,2.9hz),5.42(1h,dd,j=14.4,1.8hz),5.67(1h,d,j=6.5hz),5.72
‑
5.75(3h,m),6.83
‑
6.87(1h,m),7.01(1h,d,j=6.9hz),7.09(1h,dd,j=8.0,1.1hz),7.14
‑
7.18(1h,m),7.23(1h,d,j=7.8hz),7.37
‑
7.44
(2h,m)。
[0339]1h
‑
nmr(dmso
‑
d6)δ:2.91
‑
2.98(1h,m),3.24
‑
3.31(1h,m),3.44(1h,t,j=10.4hz),3.69(1h,dd,j=11.5,2.8hz),3.73(3h,s),4.00(1h,dd,j=10.8,2.9hz),4.06(1h,d,j=14.3hz),4.40(1h,d,j=11.8hz),4.45(1h,dd,j=9.9,2.9hz),5.42(1h,dd,j=14.4,1.8hz),5.67(1h,d,j=6.5hz),5.72
‑
5.75(3h,m),6.83
‑
6.87(1h,m),7.01(1h,d,j=6.9hz),7.09(1h,dd,j=8.0,1.1hz),7.14
‑
7.18(1h,m),7.23(1h,d,j=7.8hz),7.37
‑
7.44(2h,m)。
[0340]
实施例6:化合物33至41的制备及其非对映异构体比率
[0341]
[化学方案28]
[0342][0343]
步骤1:化合物24至32
[0344]
根据实施例5的步骤1
‑
1、1
‑
2和1
‑
3以及常规方法制备化合物24至32。
[0345]
步骤2:化合物33至41
[0346]
用实施例5中描述的步骤2的程序,使化合物24至32各自与化合物19反应,并且通过hplc分析反应混合物,以确定化合物33至41的非对映异构体比率。
[0347]
化合物33a:tr 6.4min/化合物33b:tr 6.7min
[0348]
化合物34a:tr 8.9min/化合物34b:tr 9.3min
[0349]
化合物35a:tr 9.8min/化合物35b:tr 10.1min
[0350]
化合物36a:tr 10.7min/化合物36b:tr 11.1min
[0351]
化合物37a:tr 12.5min/化合物37b:tr 12.8min
[0352]
化合物38a:tr 13.4min/化合物38b:tr 13.8min
[0353]
化合物39a:tr 8.7min/化合物39b:tr 9.0min
[0354]
化合物40a:tr 9.9min/化合物40b:tr 10.2min
[0355]
化合物41a:tr 10.6min/化合物41b:tr 11.0min
[0356]
(tr:hplc测量中的保留时间)
[0357]
(测量条件)
[0358]
柱:kinetex
tm
(2.6μm c18内径4.6
×
100mm)(岛津公司(shimadzu))
[0359]
流速:1.0ml/min;紫外检测波长:254nm;
[0360]
流动相:[a]0.1%甲酸水溶液,[b]0.1%甲酸的乙腈溶液
[0361]
梯度程序:以25%溶剂[b]开始;在10min内用25%至70%溶剂[b]的线性梯度;并且用70%溶剂[b]维持8min。
[0362]
测试实施例1:测量帽依赖性核酸内切酶(cen)抑制活性
[0363]
1)底物的制备
[0364]
购买30merrna(5'
‑
pp
‑
[m2'
‑
o]gaa uau(
‑
cy3)gca uca cua gua agc uuu gcu cua
‑
bhq2
‑3’
,japan bioservice),其中在5'端的g已被二磷酸酯修饰,在2'位的羟基已被甲氧基化修饰,在自5'端起第6位的u已用cy3标记,并且3'端已用bhq2标记,并且使用由epicentre制造的scriptcap系统添加帽结构,以得到产物m7g[5']
‑
ppp
‑
[5'][m2'
‑
o]gaa uau(
‑
cy3)gca uca cua gua agc uuu gcu cua(
‑
bhq2)
‑
3')。产物通过变性聚丙烯酰胺凝胶电泳进行分离和纯化,并且用作底物。
[0365]
2)酶的制备
[0366]
根据标准方法从病毒颗粒制备rnp(参考文献:virology(1976)73,第327
‑
338页,olga m.rochovansky)。具体地,用a/wsn/33病毒(1
×
103pfu/ml,200μl)接种10日龄的含胚鸡蛋。在37℃下孵育2天之后,回收鸡蛋的尿囊液。将病毒颗粒通过用20%蔗糖超速离心来纯化,用tritonx
‑
100和溶血卵磷脂溶解,并且通过在用30%
‑
70%甘油的密度梯度下超速离心来收集rnp级分(50%
‑
70%甘油级分),并将其用作酶溶液(含有约1nm pb1/pb2/pa复合物)。
[0367]
3)酶促反应
[0368]
将2.5μl酶促反应溶液(53mm tris
‑
盐酸盐(ph 7.8)、1mm mgcl2、1.25mm二硫苏糖醇、80mm nacl、12.5%甘油、0.15μl酶溶液)分配到384孔聚丙烯板中。然后,将0.5μl已用二甲基亚砜(dmso)连续稀释的测试化合物溶液添加到该板中。对于阳性对照(pc)和阴性对照(nc),分别将0.5μl dmso添加到该板中。将溶液充分混合。然后,添加2μl底物溶液(1.4nm底
物rna,0.05%tween 20)以引发反应。在室温下孵育60分钟之后,将1μl反应溶液添加到10μl hi
‑
di甲酰胺溶液中(含有genescan 120liz size standard作为粒度分级标记物:由应用生物系统公司(applied biosystem(abi))制造),以淬灭反应。对于nc,通过在反应开始之前预先添加edta(4.5mm)来淬灭反应(所示浓度是最终浓度)。
[0369]
4)抑制率的测量(ic
50
值)
[0370]
将如上淬火的反应溶液在85℃加热5分钟,并然后在冰上快速冷却2分钟,并在abi prizm 3730遗传分析仪(genetic analyzer)上分析。通过分析软件abi genemapper定量帽依赖性核酸内切酶产物的峰。确定测试化合物的cen反应抑制率(%),其中pc和nc的荧光强度分别为0%抑制和100%抑制,并且使用曲线拟合软件(xlfit 2.0:model 205(idbs))确定ic
50
值。
[0371]
测试实施例2:cpe抑制效果
[0372]
<材料>
[0373]
·
2%fcs e
‑
mem(通过将卡那霉素和fcs添加到mem(最低必需培养基)中来制备)(英杰公司(invitrogen)
[0374]
·
0.5%bsa e
‑
mem(通过将卡那霉素和bsa添加到mem(最低必需培养基)中来制备)(英杰公司)
[0375]
·
hbss(汉克斯平衡盐溶液(hanks'balanced salt solution))
[0376]
·
mdbk细胞(用2%fcs e
‑
mem调节至适当的细胞数(3
×
105/ml))
[0377]
·
mdck细胞(通过用hbss洗涤两次来制备并用0.5%bsa e
‑
mem调节至适当的细胞数(5
×
105/ml))
[0378]
·
胰蛋白酶溶液(将来自猪胰腺的胰蛋白酶(sigma)溶解于pbs(
‑
)中并且通过0.45μm过滤器过滤)
[0379]
·
envision(珀金埃尔默公司(perkinelmer))
[0380]
·
wst
‑
8试剂盒(岸田化学株式会社(kishida chemical co.,ltd.))
[0381]
·
10%sds溶液
[0382]
<方法>
[0383]
测试样品的稀释和分配
[0384]
作为培养基,2%fcs e
‑
mem用于mdbk细胞,并且0.5%bsa e
‑
mem用于mdck细胞。使用相同的培养基稀释病毒、细胞和测试样品。
[0385]
将测试样品用培养基初步稀释至适当浓度,并且在96孔板中制备2至5倍连续稀释系列(50μl/孔)。制备两组板分别用于抗流感活性测量和细胞毒性测量。对于每种药物,进行三重测量。
[0386]
当使用mdck细胞用于测量抗流感活性时,将胰蛋白酶添加到细胞中,使得最终浓度为3μg/ml。
[0387]
流感病毒的稀释和分配
[0388]
将流感病毒用培养基稀释至适当浓度,并且以50μl/孔分配到含有测试样品的96孔板中。向用于测量细胞毒性的板中分配50μl/孔的培养液。
[0389]
细胞的稀释和分配
[0390]
将细胞稀释至适当的细胞数,并且以100μl/孔分配至含有测试样品的96孔板中。
[0391]
将细胞培养物使用板混合器混合,并且在co2孵育箱中进行孵育。将细胞培养3天用于进行抗流感活性测量和细胞毒性测量。
[0392]
wst
‑
8的分配
[0393]
用肉眼并且在显微镜下观察培养了3天的96孔板,以检查细胞的形态和晶体的存在或不存在。除去上清液,以便不从板中吸入细胞。
[0394]
将wst
‑
8试剂盒用培养基稀释10倍,并且将100μl wst
‑
8溶液分配到每个孔中。在使用板混合器混合之后,将细胞在co2孵育箱中培养1至3小时。
[0395]
为了测量抗流感活性,在将板孵育之后,将10%sds溶液(10μl)分配到每个孔中以灭活病毒。
[0396]
吸光度的测量
[0397]
在96孔板中混合之后,在envision上在450nm/620nm的两个波长下测量吸光度。
[0398]
<测量值的计算>
[0399]
基于以下方程式,使用microsoft excel或具有等效计算和处理能力的程序计算这些值。
[0400]
计算实现50%流感感染的细胞死亡抑制的有效浓度(ec
50
)
[0401]
ec
50
=10
z
[0402]
z=(50%
‑
高%)/(高%
‑
低%)
×
{log(高浓度)
‑
log(低浓度)} log(高浓度)
[0403]
化合物(v)的测试实施例1和测试实施例2的结果如下所示。
[0404]
测试实施例1(cen ic 50):1.93nm,
[0405]
测试实施例2(cpe ec 50):1.13nm
[0406]
以上结果揭示了,具有式(v)的化合物显示出高的帽依赖性核酸内切酶(cen)抑制活性和/或高的cpe抑制作用,并且因此可用作用于治疗和/或预防由感染流感病毒诱发的症状和/或疾病的药剂。
[0407]
化合物(v)和(vi)的生物测试实施例描述如下。
[0408]
测试实施例3:cyp抑制测试
[0409]
使用可商购的合并的人类肝微粒体,并使用人类主要五种cyp酶形式(cyp1a2、2c9、2c19、2d6、3a4)的典型底物代谢反应(即,7
‑
乙氧基试卤灵o
‑
脱乙基化(cyp1a2)、甲苯磺丁脲甲基
‑
羟基化(cyp2c9)、美芬妥英4'
‑
羟基化(cyp2c19)、右美沙芬o
‑
脱甲基化(cyp2d6)和特非那定(terfenedine)羟基化(cyp3a4))作为参考,对于每种代谢物生成,评估被化合物(v)抑制的程度。
[0410]
反应条件如下:
[0411]
底物:0.5μmol/l乙氧基试卤灵(cyp1a2)、100μmol/l甲苯磺丁脲(cyp2c9)、50μmol/l s
‑
美芬妥英(cyp2c19)、5μmol/l右美沙芬(cyp2d6)、1μmol/l 特非那定(cyp3a4);反应时间:15分钟;反应温度:37℃;酶:合并的人类肝微粒体0.2mg蛋白/ml;化合物(v)的浓度:1、5、10、20μmol/l(4个点)。
[0412]
对于这五种物质中的每一种,通过按如上所述的比例将底物、人类肝微粒体和化合物(v)添加到50mmol/l hepes缓冲液中在96孔板上制备反应溶液。添加nadph(辅因子)以引发代谢反应。在37℃下孵育15分钟之后,添加甲醇/乙腈溶液(1/1(v/v))以淬灭反应。在3000rpm下离心15分钟之后,通过荧光多标记计数器测量上清液中的试卤灵(cyp1a2代谢
物),并且通过lc/ms/ms测量羟基甲苯磺丁脲(cyp2c9代谢物)、4'
‑
羟基美芬妥英(cyp2c19代谢物)、右啡烷(cyp2d6代谢物)和特非那定(terfenadine)醇(cyp3a4代谢物)。
[0413]
作为对照,将dmso(用于溶解化合物(v)的溶剂)单独添加到反应体系中。在化合物(v)的每个浓度下计算相对于对照物(100%)该化合物的剩余活性(%),并且使用浓度和抑制率通过逻辑斯谛模型(logistic model)由反向推测来计算ic
50
。
[0414]
(结果)
[0415]
化合物(v):对于这五种酶形式,>20μmol/l
[0416]
测试实施例4:ba测试
[0417]
用于口服吸收研究的材料和方法
[0418]
(1)动物:小鼠或sd大鼠
[0419]
(2)繁殖条件:允许小鼠或sd大鼠自由摄取固体饲料和灭菌自来水。
[0420]
(3)剂量和分组:以预定剂量口服或静脉内给予;分组如下(剂量取决于化合物)
[0421]
口服给予:1至30mg/kg(n=2至3)
[0422]
静脉内给予:0.5至10mg/kg(n=2至3)
[0423]
(4)给药溶液的制备:溶液或悬浮液状态,用于口服给予;溶解状态,用于静脉内给予
[0424]
(5)给予方法:使用用于口服给予的口服探针进行强制性胃内给予;使用用于静脉内给予的装有针头的注射器从臀静脉给予
[0425]
(6)终点:随时间的推移收集血液,并且通过lc/ms/ms测量化合物(v)和(vi)的血浆浓度。
[0426]
(7)统计分析:关于化合物(v)和(vi)的血浆浓度的转变,通过非线性最小二乘法程序winnonlin
tm
计算在血浆浓度
‑
时间曲线下的面积(auc),并且化合物(v)和(vi)的生物利用度(ba)由口服给予组和静脉内给予组的auc计算。
[0427]
(结果)
[0428]
化合物(v):4.2%
[0429]
化合物(vi):14.9%
[0430]
以上结果揭示了,相比母体化合物,前药具有改善的生物利用度。
[0431]
因此,具有式(vi)的化合物具有优异的口服吸收性,并且可用作用于治疗和/或预防由感染流感病毒诱发的症状和/或疾病的药剂。
[0432]
测试实施例5:代谢稳定性测试
[0433]
使化合物(v)与可商购的合并的人类肝微粒体反应一定时间。通过比较反应的样品和未反应的样品来计算化合物的剩余率,以评估化合物(v)在肝脏中的代谢程度。
[0434]
使该化合物在0.2ml含有0.5mg蛋白质/ml人类肝微粒体的缓冲液(50mmol/l tris
‑
hcl ph 7.4,150mmol/l氯化钾,10mmol/l氯化镁)中在1mmol/l nadph(氧化反应)存在下在37℃下反应0分钟或30分钟。在反应之后,将50μl反应溶液添加到100μl甲醇/乙腈=1/1(v/v)中,混合并且在3000rpm下离心15分钟。通过lc/ms/ms测量上清液中化合物(v)的量,并且计算反应后化合物的剩余率,其中在0分钟反应时间时化合物的量为100%。水解反应在不存在nadph下进行,并且葡萄糖醛酸化反应在5mmol/l udp
‑
葡萄糖醛酸代替nadph的存在下进行,并且随后的程序以与所述相同的方式进行。
[0435]
(结果)
[0436]
2μmol/l化合物在氧化代谢中的剩余率如下所示。
[0437]
化合物(v):90.1%
[0438]
测试实施例6:cyp3a4荧光mbi测试
[0439]
cyp3a4荧光mbi测试研究化合物(v)在代谢反应中对cyp3a4抑制的增强。通过cyp3a4酶(在大肠杆菌中表达的酶)使7
‑
苄氧基三氟甲基香豆素(7
‑
bfc)脱苄基,并且产生发射荧光的代谢物7
‑
羟基三氟甲基香豆素(7
‑
hfc)。使用7
‑
hfc生成反应作为指标进行该测试。
[0440]
反应条件如下:
[0441]
底物,5.6μmol/l 7
‑
bfc;预反应时间,0或30分钟;反应时间,15分钟;反应温度,25℃(室温);cyp3a4含量(在大肠杆菌中表达),预反应时为62.5pmol/ml,反应时为6.25pmol/ml(稀释10倍);化合物(v)的浓度,0.625、1.25、2.5、5、10、20μmol/l(6个点)。
[0442]
将如上所述的在k
‑
pi缓冲液(ph 7.4)中含有该酶和化合物(v)的预反应溶液添加到96孔板中。将溶液的一部分转移到另一个96孔板中,并且用底物和k
‑
pi缓冲液进行1/10稀释。添加nadph作为辅因子以引发反应(没有预孵育),并且在孵育预定时间后添加乙腈/0.5mol/l tris(三羟基氨基甲烷)=4/1(v/v)以淬灭反应。另外,向另一预孵育溶液中添加nadph以开始预孵育(有预孵育)。在预孵育预定时间之后,将溶液的一部分转移到另一个板中并用底物和k
‑
pi缓冲液进行1/10稀释以引发反应。在反应预定时间后,添加乙腈/0.5mol/l三(三羟基氨基甲烷)=4/1(v/v)以淬灭反应。对于在其上进行反应的每个板,通过荧光板读数器测量代谢物7
‑
hfc的荧光值(ex=420nm,em=535nm)。
[0443]
作为剩余活性的对照,仅将dmso(即,用于溶解化合物(v)的溶剂)添加到反应体系中,并且计算在溶液中各浓度的化合物(v)的剩余活性(%)。通过使用浓度和抑制率的逻辑斯谛模型由反向推测来计算ic
50
值。将5μm或更大的ic
50
值差异定义为( ),并且将3μm或更小的差异定义为(
‑
)。
[0444]
(结果)
[0445]
化合物(v):(
‑
)
[0446]
测试实施例7:波动埃姆斯测试
[0447]
评价化合物(v)的致突变性。
[0448]
将每种20μl冷冻储存的鼠伤寒沙门氏菌(ta98和ta100菌株)接种于10ml液体营养培养基(2.5%oxoid营养肉汤2号)中,并且将培养物在37℃下振荡孵育10小时。将ta98培养物(9ml)离心(2000
×
g,10分钟)以去除培养基,并且将细菌悬浮在9ml的micro f缓冲液(k2hpo4:3.5g/l,kh2po4:1g/l,(nh4)2so4:1g/l,柠檬酸三钠二水合物:0.25g/l,mgso4·
7h2o:0.1g/l)中。将悬浮液添加到110ml exposure培养基(含有生物素:8μg/ml、组氨酸:0.2μg/ml、葡萄糖:8mg/ml的micro f缓冲液)中。将ta100培养物(3.16ml)添加到120ml exposure培养基中以制备测试细菌溶液。将该测试细菌溶液(588μl)或在用代谢活化系统的情况下,将测试细菌溶液(498μl)和s9混合物(90μl)的混合溶液与每种12μl以下溶液混合:化合物(v)的dmso溶液,从最大剂量50mg/ml开始以数个步骤连续稀释2或3倍;dmso作为阴性对照;50μg/ml 4
‑
硝基喹啉
‑1‑
氧化物的dmso溶液作为没有代谢活化系统的ta98的阳性对照;0.25μg/ml 2
‑
(2
‑
呋喃基)
‑3‑
(5
‑
硝基
‑2‑
呋喃基)丙烯酰胺的dmso溶液作为没有代
谢活化系统的ta100的阳性对照;40μg/ml 2
‑
氨基蒽的dmso溶液作为具有代谢活化系统的ta98的阳性对照;20μg/ml 2
‑
氨基蒽的dmso溶液作为具有代谢活化系统的ta100的阳性对照。将混合物在37℃下振荡下孵育90分钟。将如此暴露于化合物(v)的细菌溶液(460μl)添加到2300μl指示剂培养基(含有生物素:8μg/ml、组氨酸:0.2μg/ml、葡萄糖:8mg/ml、溴甲酚紫:37.5μg/ml的micro f缓冲液)中,并且将每种50μl混合物分配到微量板中(每剂量48个孔)。在37℃下静置培养3天之后,含有已通过编码氨基酸(组氨酸)合成酶的基因的突变而获得增殖能力的细菌的孔的颜色由于ph变化而从紫色变为黄色。对48个孔/剂量中黄色孔的数目进行计数,以通过与阴性对照组比较来评价致突变性。(
‑
)意指致突变性为阴性,并且( )意指阳性。
[0449]
(结果)
[0450]
化合物(v):(
‑
)
[0451]
测试实施例8:herg测试
[0452]
为了评估心电图qt间期延长的风险的目的,使用表达人类ether
‑
a
‑
go
‑
go相关基因(herg)通道的hek293细胞研究化合物(v)延迟整流k 电流(ikr)的影响,延迟整流k 电流在心室复极化过程中发挥重要作用。
[0453]
使用自动膜片钳系统(patchxpress 7000a,axon instruments inc.),通过全细胞膜片钳方法将细胞维持在
‑
80mv的膜电位。记录通过在 40mv下去极化脉冲刺激2秒、以及进一步在
‑
50mv下复极化脉冲刺激2秒所诱发的ikr。在产生的电流稳定之后,含有目标浓度的化合物(v)的细胞外溶液(nacl:135mmol/l,kcl:5.4mmol/l,nah2po4:0.3mmol/l,cacl2·
2h2o:1.8mmol/l,mgcl2·
6h2o:1mmol/l,葡萄糖:10mmol/l,hepes(4
‑
(2
‑
羟乙基)
‑1‑
哌嗪乙磺酸):10mmol/l,ph=7.4)在室温下施用于细胞10分钟。从所记录的i
kr
,使用分析软件(dataxpress ver.1,分子器件公司(molecular devices corporation))基于静膜电位下的电流值确定尾峰电流的绝对值。此外,计算在施用化合物(v)之前对尾峰电流的抑制率,并且与载体施用组(0.1%二甲基亚砜溶液)比较以评估化合物(v)对i
kr
的影响。
[0454]
(结果)
[0455]
0.3至10μmol/l化合物的抑制率如下所示。
[0456]
化合物(v):7.9%
[0457]
测试实施例9:溶解度测试
[0458]
在1%dmso添加条件下测定化合物(v)的溶解度。制备10mmol/l该化合物的dmso溶液,并且将2μl化合物(v)溶液分别添加到198μl jp
‑
1溶液(氯化钠2.0g、盐酸7.0ml和水以达到1000ml)和jp
‑
2溶液(3.40g磷酸二氢钾和3.55g无水磷酸氢二钠溶于以使得达到1000ml的水中,然后将其1体积添加到1体积水中)。在室温下振荡1小时之后,过滤混合物。将滤液用甲醇/水=1/1(v/v)稀释10倍,并且使用lc/ms通过绝对校准方法测量滤液中化合物的浓度。
[0459]
(结果)
[0460]
化合物(v):42.2μmol/l
[0461]
测试实施例10:粉末溶解度测试
[0462]
将适量的化合物(v)放入适当的容器中。向对应容器中添加200μl jp
‑
1溶液(氯化钠2.0g、盐酸7.0ml和以使得达到1000ml的水)、200μl jp
‑
2溶液(将500ml水添加到50ml磷
酸盐缓冲液(ph 6.8)中)和200μl的20mmol/l牛磺胆酸钠(tca)/jp
‑
2溶液(tca 1.08g和以使得达到100ml的水)。如果化合物(v)在添加到测试溶液中之后完全溶解,则适当地进一步添加化合物(v)。将容器密封,并且在37℃下振荡1小时。过滤混合物,并且向每个滤液(100μl)中添加100μl甲醇,使得滤液被稀释2倍。必要时改变稀释比。观察稀释物的气泡和沉淀物,并且然后将容器密封并振荡。化合物(v)的定量通过hplc用绝对校准方法进行。
[0463]
(结果)
[0464]
化合物(v):jp
‑
1溶液7.1μg/ml,jp
‑
2溶液4.4μg/ml,20mmol/l tca/jp
‑
2溶液16.1μg/ml
[0465]
测试实施例11埃姆斯测试
[0466]
使用沙氏杆菌(鼠伤寒沙门氏菌(salmonella typhimurium))ta 98、ta100、ta1535和ta1537以及大肠杆菌wp2uvra作为测试菌株在预孵育中在有或没有代谢活化的情况下进行埃姆斯测试,以检查化合物(v)的基因致突变性的存在或不存在。
[0467]
(结果)
[0468]
化合物(v):(
‑
)
[0469]
测试实施例12:光致溶血测试
[0470]
将化合物(v)以预定浓度溶解,并且在微量板上与由绵羊的绵羊去纤维蛋白血液制备的0.1%至0.0008%红血球悬浮液(2.5v/v%)混合。使用紫外荧光灯(gl20se灯,sankyo denki和fl20s
‑
blb灯,松下公司(panasonic))进行在uva和uvb波长区域中的光照射(10j/cm2,290
‑
400nm)。收集并离心照射后的混合溶液。在离心之后,将上清液收集并且转移到微量板中,并且测量该上清液的吸光度(在540nm或630nm处)。将在540和630nm处的吸光度分别用作生物膜损伤(%光溶血)和脂质膜过氧化(高铁血红蛋白产生)的指标。(
‑
):光致溶血率小于10%,并且在630nm处的吸光度变化小于0.05;( ):光致溶血率为10%或更大,并且在630nm处的吸光度变化为0.05或更大。
[0471]
(结果)
[0472]
化合物(v):(
‑
)
[0473]
图7和图8示出了在非禁食条件下将化合物(vi)口服给予大鼠之后化合物(v)及其前药化合物(vi)在血浆中的浓度的时程。
[0474]
化合物(vi)在血浆样品中的浓度低于定量极限,表明作为化合物(v)的前药的化合物(vi)在给予之后迅速在体内转化为化合物(v)(参见图8)。
[0475]
这些测试结果揭示了,前药化合物在口服给予之后被吸收到体内,并在血液中迅速转化为其母体化合物。因此,化合物(v)和(vi)可用作用于治疗和/或预防由感染流感病毒诱发的症状和/或疾病的药剂。
[0476]
测试实施例13:静脉内给予测试
[0477]
材料和方法
[0478]
(1)测试动物:sd大鼠
[0479]
(2)饲养条件:允许sd大鼠自由获取固体饲料和无菌自来水。
[0480]
(3)剂量和分组设置:根据预定剂量静脉内给予。如下设定组(可以针对每种化合物改变剂量)。
[0481]
静脉内给予:0.5至1mg/kg(n=2至3)
[0482]
(4)给予液的制备:溶解用于静脉内给予。
[0483]
(5)给予方法:用装有针头的注射器从尾静脉给予。
[0484]
(6)终点:随时间的推移收集血液,并且通过lc/ms/ms测量化合物(v)的血浆浓度
[0485]
(7)统计分析:使用非线性最小二乘法程序winnonlin
tm
由化合物(v)在血浆中的浓度的时程计算全身清除率(cltot)和消除半衰期(t1/2,z)。
[0486]
(结果)
[0487]
化合物(v):
[0488]
cltot:16.4ml/min/kg
[0489]
t1/2,z:3.4小时
[0490]
以上结果揭示了,化合物(v)具有低的全身清除率和长的半衰期。
[0491]
因此,化合物(v)可以是持久性优异并且可用作用于治疗和/或预防由感染流感病毒诱发的症状和/或疾病的药剂的药物。
[0492]
本发明的化合物和方法可用作用于生产有用化合物的中间体,该有用化合物作为用于治疗和/或预防由感染流感病毒诱发的症状和/或疾病的药剂。根据本发明的方法,可以有效率地生产具有式(v)的化合物和具有式(vi)的化合物。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。