一种
α
‑
溴代苯乙酮类化合物的制备方法
技术领域
1.本发明属于有机合成技术领域,具体涉及一种α
‑
溴代苯乙酮类化合物的制备方法。
背景技术:
2.α
‑
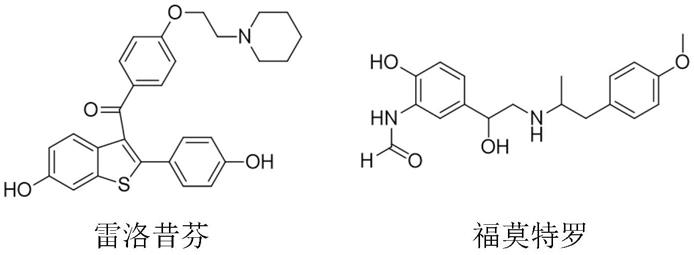
溴代苯乙酮类化合物及其衍生物是重要的医药、农药中间体,如:对甲氧基溴代苯乙酮是治疗和预防妇女骨质疏松症药物雷洛昔芬的关键中间体;3
‑
硝基
‑4‑
苄氧基
‑
α
‑
溴代苯乙酮类化合物是治疗哮喘药物福莫特罗的关键中间体。
[0003][0004]
现有技术中,制备α
‑
溴代苯乙酮类化合物的方法主要包括:
[0005]
1)苯乙酮与br2、nbs、cubr2等溴化试剂的溴代反应(j.heterocyclic chem.,2020,57,1
‑
9;eur.j.org.chem.,2017,2017,781
‑
785;eur.j.med.chem.,2015,18
‑
23.),这是目前制备α
‑
溴代苯乙酮类化合物最常用的方法;
[0006]
2)利用氧化剂氧化溴负离子原位形成溴,然后再与苯乙酮进行溴代反应(tetrahedron lett.,2012,53,191
‑
195.);
[0007]
3)通过(2
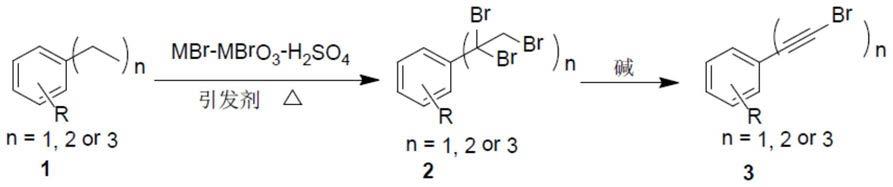
‑
溴乙炔基)苯的水合反应(j.org.chem.,2013,78,9190
‑
9195;chin.j.chem.,2016,34,1251
‑
1254;tetrahedron lett.,2016,57,4983
‑
4986.)。
[0008]
上述α
‑
溴代苯乙酮类化合物的现有制备技术具有如下不足之处:
[0009]
1)乙酰基是属于间位定位基,对苯环具有钝化作用,不利于在苯环上引入其它取代基,因此要想得到苯环上带有其它不同取代基的苯乙酮,具有一定的难度;
[0010]
2)br2、nbs、cubr2等溴化试剂具有污染大、成本高等特点;
[0011]
3)(2
‑
溴乙炔基)苯的合成路线比较复杂,部分(2
‑
溴乙炔基)苯衍生物很不稳定,因此原料来源也受限。
技术实现要素:
[0012]
基于现有技术中存在的上述缺点和不足,本发明的目的之一是至少解决现有技术中存在的上述问题之一或多个,换言之,本发明的目的之一是提供满足前述需求之一或多个的一种α
‑
溴代苯乙酮类化合物的制备方法。
[0013]
为了达到上述发明目的,本发明采用以下技术方案:
[0014]
一种α
‑
溴代苯乙酮类化合物的制备方法,包括以下步骤:
[0015]
(1)在有机溶剂中,以溴酸盐、溴化盐及硫酸作为溴化试剂,并在引发剂的作用下,对乙基苯类化合物进行自由基溴代反应,得到(1,1,2
‑
三溴乙基)苯衍生物;
[0016]
(2)在酸性溶液中,(1,1,2
‑
三溴乙基)苯衍生物进行水解反应,得到α
‑
溴代苯乙酮类化合物;
[0017]
其中,乙基苯类化合物的化学式为:其中,n为乙基的个数,取值为1、2或3;r为氢原子或取代基;
[0018]
(1,1,2
‑
三溴乙基)苯衍生物的化学式为:
[0019]
α
‑
溴代苯乙酮类化合物的化学式为:
[0020]
作为优选方案,所述步骤(1)中,乙基苯类化合物、溴酸盐、溴化盐及硫酸的物质的量之比为1.0:(2.0~2.4)
×
n:(1.0~1.2)
×
n:(1.5~1.8)
×
n;
[0021]
所述有机溶剂的体积与乙基苯类化合物的物质的量之比为1.6~4ml/mmol;
[0022]
所述引发剂的物质的量与乙基苯类化合物的物质的量之比为(0.04~0.12)
×
n mol/mmol。
[0023]
作为优选方案,所述步骤(1)中还加入水,水的体积与乙基苯类化合物的物质的量之比为1.6~4ml/mmol。
[0024]
作为优选方案,所述有机溶剂为二氯甲烷、1,2
‑
二氯乙烷、四氯化碳中的一种或几种的混合。
[0025]
作为优选方案,所述溴化物为溴化钠、溴化钾中的一种或两种的混合;所述溴酸盐为溴酸钠、溴酸钾中的一种或两种的混合。
[0026]
作为优选方案,所述引发剂为偶氮二异庚腈、偶氮二异丁腈、过氧化二苯甲酰中的一种或几种的混合。
[0027]
作为优选方案,所述步骤(2)中,酸性溶液的体积与(1,1,2
‑
三溴乙基)苯衍生物的物质的量之比为1~6ml/mmol。
[0028]
作为优选方案,所述酸性溶液为氢溴酸、盐酸、硫酸中的一种或几种的混合。
[0029]
作为优选方案,所述酸性溶液的质量百分比浓度为10~40%。
[0030]
作为优选方案,所述取代基为硝基、卤素原子、叔丁基、co2me或ococh3。
[0031]
本发明与现有技术相比,有益效果是:
[0032]
(1)本发明的α
‑
溴代苯乙酮类化合物的制备方法,采用两步法即可制得α
‑
溴代苯乙酮类化合物,工艺简单;
[0033]
(2)反应选择性好,产品收率高;
[0034]
(3)所有原料均为常用化学品,价廉易得,合成成本低;
[0035]
(4)采取的操作均为常规操作,简单安全;
[0036]
(5)水解反应所用的氢溴酸可以重复利用,水解下来的溴原子不会损耗;
[0037]
(6)本发明的底物适用范围广。
附图说明
[0038]
图1是本发明实施例的α
‑
溴代苯乙酮类化合物的制备方法的流程图。
具体实施方式
[0039]
以下通过具体实施例对本发明的技术方案作进一步解释说明。
[0040]
如图1所示,本发明的α
‑
溴代苯乙酮类化合物的制备方法,从价廉易得的乙基苯类化合物1出发,以mbr
‑
mbro3‑
h2so4(m=na or k)为溴化试剂,并在引发剂的作用下,通过自由基溴代反应制备得到中间体2,即(1,1,2
‑
三溴乙基)苯衍生物;
[0041]
中间体2在酸性水溶液中水解,得到α
‑
溴代苯乙酮类化合物3。
[0042]
其中,乙基苯类化合物的化学式为:其中,n为乙基的个数,取值为1、2或3;r为氢原子或取代基;取代基为硝基、卤素原子、叔丁基等取代基,还可以为co2me、ococh3等取代基,但这类基团在水解过程中可以同时发生水解,得到cooh、oh等重要官能团;
[0043]
(1,1,2
‑
三溴乙基)苯衍生物的化学式为:
[0044]
α
‑
溴代苯乙酮类化合物的化学式为:
[0045]
本发明的α
‑
溴代苯乙酮类化合物的制备方法,具有原料价廉易得、操作简单安全、反应选择性好、产品收率高、三废排放少等优点。以下通过具体实施例进行示例说明:
[0046]
实施例1:
[0047]
本实施例的α
‑
溴代苯乙酮的制备方法,包括以下步骤:
[0048]
(1)在25ml的三口烧瓶中分别加入乙基苯(3mmol),nabr(6.6mmol),nabro3(3.3mmol),1,2
‑
二氯乙烷(3.5ml)和水(0.4ml),然后装上尾气吸收装置和回流冷凝管,搅拌加热至回流,滴加硫酸溶液(4.95mmol)和偶氮二异丁腈溶液(0.12mmol aibn,1,2
‑
二氯乙烷为溶剂),滴加完成后,继续回流反应,用薄层色谱法跟踪检测,反应完全后,停止加热,并降至室温,加入饱和碳酸氢钠水溶液中和,用1,2
‑
二氯乙烷萃取水相,合并有机相,有机相用无水硫酸钠干燥、过滤,减压蒸馏回收有机有机溶剂,残余物经硅胶柱色谱纯化,得到中间体(1,1,2
‑
三溴乙基)苯,黄色油状物,产量1.01g,产率98%。
[0049]
其中,中间体(1,1,2
‑
三溴乙基)苯的化学式为:核磁数据如下:
[0050]1h nmr(600mhz,cdcl3)δ7.76(d,j=7.8hz,2h),7.39(t,j=7.5hz,2h),7.36(d,j=7.1hz,1h),4.66(s,2h);
13
c nmr(151mhz,cdcl3)δ141.1,129.5,128.3,127.1,64.6,45.5.gc
‑
ms(ei):calcd for c8h7br2(m
‑
br):262.9.found:262.9。
[0051]
(2)将1mmol(1,1,2
‑
三溴乙基)苯、4ml 40%氢溴酸加入到25ml三口烧瓶中,搅拌加热至105℃反应,薄层色谱法跟踪,反应完成后停止加热并降至室温,加入10ml水,搅拌均匀,静置分液,水相分别用5ml乙酸乙酯萃取3次,合并有机相用无水硫酸钠干燥,减压蒸馏回收溶剂,经硅胶柱色谱纯化,得到产品α
‑
溴代苯乙酮类化合物,白色固体,产量175mg,收率88%。
[0052]
其中,α
‑
溴代苯乙酮类化合物的化学式为:核磁数据如下:
[0053]1h nmr(600mhz,cdcl3)δ7.98(dd,j=8.4,1.2hz,2h),7.62
–
7.59(m,1h),7.51
–
7.47(m,2h),4.46(s,2h);
13
c nmr(151mhz,cdcl3)δ191.2,133.8,128.8,30.9。
[0054]
实施例2:
[0055]
本实施例的α
‑
溴代苯乙酮类化合物的制备方法与实施例1的不同之处在于:
[0056]
以3mmol 1
‑
叔丁基
‑3‑
乙基苯为原料,其他步骤参照实施例1的步骤(1),得到中间体1
‑
(1,1,2
‑
三溴乙基)
‑3‑
叔丁基苯,无色油状物,产量0.92g,产率77%。
[0057]
其中,中间体1
‑
(1,1,2
‑
三溴乙基)
‑3‑
叔丁基苯的化学式为:核磁数据如下:
[0058]1h nmr(600mhz,cdcl3)δ7.80(t,j=2.0hz,1h),7.59
‑
7.58(m,1h),7.40
‑
7.38(m,1h),7.32(t,j=7.8hz,1h),4.67(s,2h),1.37(s,9h);
13
c nmr(151mhz,cdcl3)δ151.3,140.9,128.1,126.7,124.6,124.4,65.6,45.7,35.0,31.4。
[0059]
以1mmol 1
‑
(1,1,2
‑
三溴乙基)
‑3‑
叔丁基苯为原料,其他步骤参照实施例1的步骤(2),得到产物2
‑
溴
‑1‑
(3
‑
叔丁基苯基)乙酮,无色油状物,产量173mg,产率68%。
[0060]
其中,2
‑
溴
‑1‑
(3
‑
叔丁基苯基)乙酮的化学式为:核磁数据如下:
[0061]1h nmr(600mhz,cdcl3)δ8.03(s,1h),7.79(d,j=7.7hz,1h),7.65(d,j=7.8hz,1h),7.43(t,j=7.8hz,1h),4.47(s,2h),1.36(s,9h).
13
c nmr(151mhz,cdcl3)δ191.6,152.0,133.7,131.1,128.5,126.2,125.7,34.8,31.1。
nmr(151mhz,cdcl3)δ190.1,140.5,132.2,130.3,129.2,30.3。
[0078]
实施例5:
[0079]
本实施例的α
‑
溴代苯乙酮类化合物的制备方法与实施例1的不同之处在于:
[0080]
以3mmol 1
‑
乙基
‑4‑
硝基苯为原料,其他步骤参照实施例1的步骤(1),得到中间体1
‑
(1,1,2
‑
三溴乙基)
‑4‑
硝基苯,黄色固体,产量0.52g,产率45%。
[0081]
其中,中间体1
‑
(1,1,2
‑
三溴乙基)
‑4‑
硝基苯的化学式为:核磁数据如下:
[0082]1h nmr(600mhz,cdcl3)δ8.26
–
8.23(m,2h),7.95
–
7.93(m,2h),4.64(s,2h);
13
c nmr(151mhz,cdcl3)δ148.0,147.3,128.5,123.4,61.1,44.6.gc
‑
ms(ei):calcd for c8h6br2no2(m
‑
br):307.9.found:307.9。
[0083]
以1mmol 1
‑
(1,1,2
‑
三溴乙基)
‑4‑
硝基苯为原料,40%氢溴酸用量为5ml,其他步骤参照实施例1的步骤(2),得到产物2
‑
溴
‑1‑
(4
‑
硝基苯基)乙酮,黄色固体,产量180mg,产率74%。
[0084]
其中,2
‑
溴
‑1‑
(4
‑
硝基苯基)乙酮的化学式为:核磁数据如下:
[0085]1h nmr(600mhz,cdcl3)δ8.36
–
8.33(m,2h),8.17
–
8.14(m,2h),4.46(s,2h);
13
c nmr(151mhz,cdcl3)δ189.8,150.6,138.3,130.0,124.0,30.1。
[0086]
实施例6:
[0087]
本实施例的α
‑
溴代苯乙酮类化合物的制备方法与实施例1的不同之处在于:
[0088]
以3mmol 4
‑
乙基苯甲酸甲酯为原料,其他步骤参照实施例1的步骤(1),得到中间体4
‑
(1,1,2
‑
三溴乙基)苯甲酸甲酯,白色固体,产量0.96g,产率80%。
[0089]
其中,中间体4
‑
(1,1,2
‑
三溴乙基)苯甲酸甲酯的化学式为:核磁数据如下:
[0090]1h nmr(600mhz,cdcl3)δ8.06
–
8.03(m,2h),7.84
–
7.81(m,2h),4.64(s,2h),3.93(s,3h);
13
c nmr(151mhz,cdcl3)δ165.9,145.3,131.0,129.5,127.3,63.0,52.3,45.1。
[0091]
以1mmol 4
‑
(1,1,2
‑
三溴乙基)苯甲酸甲酯为原料,其他步骤参照实施例1的步骤(2),得到产物4
‑
(2
‑
溴乙酰基)苯甲酸,白色固体,产量204mg,产率84%。
[0092]
其中,4
‑
(2
‑
溴乙酰基)苯甲酸的化学式为:核磁数据如下:
[0093]1h nmr(600mhz,d6‑
dmso)δ8.08(q,j=8.4hz,4h),4.99(s,2h).
13
c nmr(151mhz,d6‑
dmso)δ191.6,166.6,137.2,135.1,129.7,129.0,34.5。
[0094]
实施例7:
[0095]
本实施例的α
‑
溴代苯乙酮类化合物的制备方法与实施例1的不同之处在于:
[0096]
以3mmol乙酸(4
‑
乙基苯)酯为原料,其他步骤参照实施例1的步骤(1),得到中间体乙酸[4
‑
(1,1,2
‑
三溴乙基)苯]酯,无色油状物,产量0.97g,产率81%。
[0097]
其中,中间体乙酸[4
‑
(1,1,2
‑
三溴乙基)苯]酯的化学式为:核磁数据如下:
[0098]1h nmr(600mhz,cdcl3)δ7.77(d,j=8.8hz,2h),7.12(d,j=8.8hz,2h),4.62(s,2h),2.31(s,3h).
13
c nmr(151mhz,cdcl3)δ168.8,151.1,138.5,128.5,121.2,63.4,45.4,21.1。
[0099]
以1mmol乙酸[4
‑
(1,1,2
‑
三溴乙基)苯]酯为原料,用50%硫酸代替氢溴酸,其他步骤参照实施例1的步骤(2),得到产物2
‑
溴
‑1‑
(4
‑
羟基苯基)乙酮,无色油状物,产量174mg,产率81%。
[0100]
其中,2
‑
溴
‑1‑
(4
‑
羟基苯基)乙酮的化学式为:核磁数据如下:
[0101]1h nmr(600mhz,d6‑
dmso)δ8.08(q,j=8.4hz,4h),4.99(s,2h).
13
c nmr(151mhz,d6‑
dmso)δ191.6,166.6,137.2,135.1,129.7,34.5。
[0102]
实施例8:
[0103]
本实施例的α
‑
溴代苯乙酮类化合物的制备方法与实施例1的不同之处在于:
[0104]
以3mmol 1,4
‑
二乙基苯为原料,nabr、nabro3、h2so4和aibn用量分别为13.2mmol、6.6mmol、9.9mmol和0.24mmol,其他步骤参照实施例1的步骤(1),得到中间体1,4
‑
二(1,1,2
‑
三溴乙基)苯,白色固体,产量1.64g,产率90%。
[0105]
其中,中间体1,4
‑
二(1,1,2
‑
三溴乙基)苯的化学式为:核磁数据如下:
[0106]1h nmr(600mhz,cdcl3)δ7.76(s,4h),4.62(s,4h);
13
c nmr(151mhz,cdcl3)δ142.2,127.2,62.8,45.0。
[0107]
以1mmol 1,4
‑
二(1,1,2
‑
三溴乙基)苯为原料,
t
buok和
t
buoh用量分别为4.1mmol和6ml,其他步骤参照实施例1的步骤(2),得到产物1,4
‑
二(2
‑
溴乙酰基)苯,黄色固体,产量96mg,产率30%。
[0108]
其中,1,4
‑
二(2
‑
溴乙酰基)苯的化学式为:核磁数据如下:
[0109]
1h nmr(600mhz,d6
‑
dmso)δ8.13(s,4h),5.01(s,4h);13c nmr(151mhz,d6
‑
dmso)δ191.9,138.0,129.4,34.8。
[0110]
鉴于本发明方案实施例众多,各组分的材料选择以及添加量均可在相应的范围内选取,各实施例实验数据庞大众多,不适合于此处逐一列举说明,但是各实施例所需要验证的内容和得到的最终结论均接近。故而此处不对各个实施例的验证内容进行逐一说明,仅以实施例1
‑
8作为代表说明本发明申请的优异之处。
[0111]
以上所述仅是对本发明的优选实施例及原理进行了详细说明,对本领域的普通技术人员而言,依据本发明提供的思想,在具体实施方式上会有改变之处,而这些改变也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。