1.本发明涉及精细化工技术领域,更具体地说,是涉及一种用于聚酮聚合催化剂的配体及其制备方法。
背景技术:
2.聚酮(pok)由一氧化碳、烯烃(乙烯、丙烯、苯乙烯)合成的新型绿色聚合物材料,聚酮具有光降解和生物降解特性,并且可进一步化学修饰,其优异和宽泛的性能表现,使它成为一款“天然”的热塑性工程塑料。聚酮的合成主要经历了三种催化体系的发展:自由基引发体系、γ-射线诱导引发体系和第八族过渡金属引发体系,发展至今,占主要地位的是第八族过渡金属催化体系,该体系有过渡金属化合物、双齿配体、强酸及其阴离子、氧化剂、助催化剂和溶剂等六部分组成。最初,shell公司开发的醋酸钯/1,3-双[双(2-甲氧基苯基)膦基]丙烷/三氟乙酸催化体系对乙烯和一氧化碳聚酮有较高的催化活性,而聚合催化体系中新型双齿配体的开发对提高聚酮聚合催化活性具有重要的作用。此后,st制药株式会社相继开发了2,2-二甲基-1,3-双[二(2-甲氧基苯基)膦基]丙烷、3,3-双[双-(2-甲氧基苯基)膦基甲基]-1,5-二氧杂-螺[5,5]十一烷和((2,2-二甲基-1,3-二噁烷-5,5-二基)双(亚甲基))双(双(2-甲氧基苯基)膦)等双齿配体,这些配体在聚酮合成的过程均表现出相比1,3-双[双(2-甲氧基苯基)膦基]丙烷更高的催化活性。但是这些配体合成路线长,收率低,结构复杂。
[0003]
因此,开发一种新型聚酮催化剂的配体及其制备方法,实现合成路线简单、收率高、成本低,配体结构新颖、催化活性高且具有更简单的结构和更低的分子量,成为本领域技术人员亟待解决的技术难题。
技术实现要素:
[0004]
有鉴于此,本发明的目的在于提供一种新型用于聚酮聚合催化剂的配体及其制备方法,该配体结构新颖、催化活性高且具有更简单的结构和更低的分子量,同时该制备方法工艺路线简单、成本低,产品收率高、品质好,有利于聚酮工业化。
[0005]
本发明提供了一种用于聚酮聚合催化剂的配体,具有式(i)所示的结构:
[0006][0007]
本发明还提供了一种上述技术方案所述的用于聚酮聚合催化剂的配体的制备方法,包括以下步骤:
[0008]
a)将双(2-甲氧基苯基)氧化膦与1,1
’‑
二溴二甲醚在溶剂中反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚;
[0009]
b)将步骤a)得到的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚在溶剂中进行还原反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚。
[0010]
优选的,步骤a)中所述反应在拔氢试剂的作用下进行;所述拔氢试剂包括叔丁醇钾、叔丁醇钠、氢化钠和正丁基锂中的一种或多种。
[0011]
优选的,步骤a)中所述双(2-甲氧基苯基)氧化膦、1,1
’‑
二溴二甲醚和拔氢试剂的摩尔比为(2-4):1:(2~6)。
[0012]
优选的,所述步骤a)具体为:
[0013]
a1)将双(2-甲氧基苯基)氧化膦加入到溶剂中,搅拌至完全溶解,得到双(2-甲氧基苯基)氧化膦溶液;所述的搅拌速度为100r/min~400r/min;
[0014]
a2)在步骤a1)得到的双(2-甲氧基苯基)氧化膦溶液中加入拔氢试剂进行反应,得到双(2-甲氧基苯基)氧化膦盐反应液;
[0015]
a3)在步骤a2)得到的双(2-甲氧基苯基)氧化膦盐反应液中加入1,1
’‑
二溴二甲醚进行反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚。
[0016]
优选的,步骤a3)中所述加入1,1
’‑
二溴二甲醚的温度控制为-40℃~-10℃,加入完成后先保持温度不变进行反应1h~2h,在升温至20℃~35℃反应14h~20h。
[0017]
优选的,步骤b)中所述还原反应在还原剂和三乙胺存在下进行;所述还原剂包括hsicl3、四氢铝锂和硼氢化钠中的一种或多种。
[0018]
优选的,步骤b)中所述1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚、三乙胺和还原剂的摩尔比为1:(6~10):(6~10)。
[0019]
优选的,所述步骤b)具体为:
[0020]
b1)将1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚、三乙胺和溶剂混合,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚混合液;
[0021]
b2)在步骤b1)得到的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚混合液中加入还原剂进行还原反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚。
[0022]
优选的,步骤b2)中所述加入还原剂的温度控制为0℃~40℃,加入完成后升温至回流状态,并保温反应4h~10h。
[0023]
本发明提供了一种用于聚酮聚合催化剂的配体及其制备方法;该制备方法包括以下步骤:a)将双(2-甲氧基苯基)氧化膦与1,1
’‑
二溴二甲醚在溶剂中反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚;b)将步骤a)得到的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚在溶剂中进行还原反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚。与现有技术相比,本发明选择特定原料按照特定工艺步骤,实现整体较好的相互作用,制备得到用于聚酮聚合催化剂的配体;该配体结构新颖、催化活性高且具有更简单的结构和更低的分子量;同时该制备方法工艺路线简单、成本低,产品收率高、品质好,有利于聚酮工业化。实验结果表明,采用本发明提供的制备方法产品纯度>98%,收率>85%,同时利用该配体开展聚合反应,催化活性高达25.37kg/(g-pd
·
h)。
附图说明
[0024]
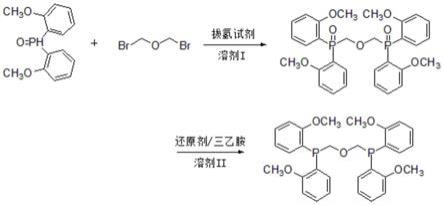
图1为本发明实施例提供的聚酮配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚的制备工艺路线图;
[0025]
图2为本发明实施例提供的聚酮配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚的
31
p nmr;
[0026]
图3为本发明实施例提供的聚酮配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚的1hnmr。
具体实施方式
[0027]
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0028]
本发明提供了一种用于聚酮聚合催化剂的配体,具有式(i)所示的结构:
[0029][0030]
该配体可命名为1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚,其结构新颖、催化活性高且具有更简单的结构和更低的分子量。实验结果表明,该配体开展聚合反应,催化活性高达25.37kg/(g-pd
·
h),满足市场生产聚酮的技术要求。
[0031]
本发明还提供了一种上述技术方案所述的用于聚酮聚合催化剂的配体的制备方法,包括以下步骤:
[0032]
a)将双(2-甲氧基苯基)氧化膦与1,1
’‑
二溴二甲醚在溶剂中反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚;
[0033]
b)将步骤a)得到的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚在溶剂中进行还原反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚。
[0034]
本发明首先将双(2-甲氧基苯基)氧化膦与1,1
’‑
二溴二甲醚在溶剂中反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚。
[0035]
在本发明中,所述双(2-甲氧基苯基)氧化膦具有下式结构:
[0036][0037]
所述1,1
’‑
二溴二甲醚具有下式结构:
[0038][0039]
本发明对所述双(2-甲氧基苯基)氧化膦和1,1
’‑
二溴二甲醚的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
[0040]
在本发明中,所述溶剂优选为四氢呋喃或乙醚;本发明对其来源没有特殊限制。
[0041]
在本发明中,所述反应优选在拔氢试剂的作用下进行;所述拔氢试剂优选包括叔丁醇钾、叔丁醇钠、氢化钠和正丁基锂中的一种或多种,更优选为叔丁醇钾、叔丁醇钠、氢化钠或正丁基锂。本发明对所述拔氢试剂的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
[0042]
在本发明中,所述双(2-甲氧基苯基)氧化膦、1,1
’‑
二溴二甲醚和拔氢试剂的摩尔比优选为(2-4):1:(2~6)。
[0043]
在此基础上,所述步骤a)优选具体为:
[0044]
a1)将双(2-甲氧基苯基)氧化膦加入到溶剂中,搅拌至完全溶解,得到双(2-甲氧基苯基)氧化膦溶液;
[0045]
a2)在步骤a1)得到的双(2-甲氧基苯基)氧化膦溶液中加入拔氢试剂进行反应,得到双(2-甲氧基苯基)氧化膦盐反应液;
[0046]
a3)在步骤a2)得到的双(2-甲氧基苯基)氧化膦盐反应液中加入1,1
’‑
二溴二甲醚进行反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚。
[0047]
在本发明中,所述的搅拌速度优选为100r/min~400r/min,更优选为200r/min~300r/min。
[0048]
在本发明中,为了避免反应过于剧烈,所述加入拔氢试剂的温度优选控制为-40℃~-5℃,加入完成后反应温度允许升温至20℃~35℃,反应时间为1h~3h。
[0049]
在本发明中,了避免反应过于剧烈以及控制杂质,所述加入1,1
’‑
二溴二甲醚的温度优选控制为-40℃~-10℃,加入完成后先保持温度不变进行反应1h~2h,在升温至20℃~35℃反应14h~20h;跟踪检测反应完毕。
[0050]
在本发明中,所述跟踪检测反应完毕后,优选还包括:
[0051]
在搅拌下将反应液缓慢滴加入20ml~40ml水中淬灭反应,滴加时间为20min~30min,充分搅拌后,分液,水洗,干燥,减压除去溶剂后,乙醇和石油醚重结晶,烘干后得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚。
[0052]
得到所述1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚后,本发明将得到的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚在溶剂中进行还原反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚。
[0053]
在本发明中,所述溶剂优选为乙腈、甲苯或二甲苯;本发明对其来源没有特殊限制。
[0054]
在本发明中,所述还原反应优选在还原剂和三乙胺存在下进行;所述还原剂优选包括hsicl3、四氢铝锂和硼氢化钠中的一种或多种,更优选为hsicl3、四氢铝锂或硼氢化钠。本发明对所述还原剂和三乙胺的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
[0055]
在本发明中,所述1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚、三乙胺和还原剂的摩尔比优选为1:(6~10):(6~10)。
[0056]
在此基础上,所述步骤b)优选具体为:
[0057]
b1)将1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚、三乙胺和溶剂混合,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚混合液;
[0058]
b2)在步骤b1)得到的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚混合液中加入还
原剂进行还原反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚。
[0059]
在本发明中,所述加入还原剂的温度优选控制为0℃~40℃,加入完成后升温至回流状态,并保温反应4h~10h;跟踪检测反应完全。
[0060]
在本发明中,所述跟踪检测反应完毕后,优选还包括:
[0061]
冷却,加6倍~10倍hsicl3摩尔当量20wt%~40wt%氢氧化钠水溶液淬灭反应,静置分液,分出有机相,乙腈萃取两次,合并有机相,饱和食盐水水洗一次,干燥,减压除去溶剂,甲醇重结晶,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚。
[0062]
本发明提供了一种新型聚酮催化剂的配体的制备方法,以双(2-甲氧基苯基)氧化膦为起始原料,先与1,1
’‑
二溴二甲醚进行反应得到中间产物得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚,再经过还原合成1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚;该制备方法工艺路线简单,产品收率高、品质好,配体结构新颖,利用该配体开展聚合反应,催化活性高达25.37kg/(g-pd
·
h),该配体具有更简单的结构和更低的分子量,这降低了聚酮生产成本,更有利于聚酮工业化。
[0063]
本发明提供了一种用于聚酮聚合催化剂的配体及其制备方法;该制备方法包括以下步骤:a)将双(2-甲氧基苯基)氧化膦与1,1
’‑
二溴二甲醚在溶剂中反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚;b)将步骤a)得到的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚在溶剂中进行还原反应,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚。与现有技术相比,本发明选择特定原料按照特定工艺步骤,实现整体较好的相互作用,制备得到用于聚酮聚合催化剂的配体;该配体结构新颖、催化活性高且具有更简单的结构和更低的分子量;同时该制备方法工艺路线简单、成本低,产品收率高、品质好,有利于聚酮工业化。实验结果表明,采用本发明提供的制备方法产品纯度>98%,收率>85%,同时利用该配体开展聚合反应,催化活性高达25.37kg/(g-pd
·
h)。
[0064]
为了进一步说明本发明,下面通过以下实施例进行详细说明。本发明以下实施例中所用的原料均为市售商品。
[0065]
实施例1
[0066]
一种新型聚酮聚合催化剂的配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚的制备方法,工艺路线如图1所示,图1为本发明实施例提供的聚酮配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚的制备工艺路线图;其具体步骤为:
[0067]
(1)1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚的制备:
[0068]
无水无氧条件下,将双(2-甲氧基苯基)氧化膦2.35g加入30ml四氢呋喃中,搅拌至溶解,搅拌速度为200r/min,降温至-20℃,开始控温滴加11.2ml 1.6m/ln-buli的溶液,20min后滴加完毕,缓慢升温至30℃搅拌1h后(搅拌速度不变),降温至-20℃,开始控温滴加1,1
’‑
二溴二甲醚0.61g,20min后滴加完毕,保温反应1h后,升温至30℃,保温反应15h后,跟踪检测反应完毕,在搅拌下将反应液缓慢滴加入30ml水中,滴加时间为25min,充分搅拌后,分液,水洗,干燥,减压除去溶剂后,乙醇和石油醚重结晶,烘干后得到白色的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚1.60g,纯度98.5%,收率93.14%。
[0069]
(2)1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚:
[0070]
无水无氧条件下,将1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚2.60g和三乙胺3.62g加入20ml乙腈中,控温20℃,滴加hsicl
3 4.88g,15min滴加完毕后,升温至回流状态,
反应6h后,跟踪检测反应完全,冷却,加8倍hsicl3摩尔当量30wt%氢氧化钠水溶液淬灭反应,静置分液,分出有机相,乙腈萃取两次,合并有机相,饱和食盐水水洗一次,干燥,减压除去溶剂,甲醇重结晶,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚2.18g,纯度99.23%,收率90.13%。
[0071]
实施例2
[0072]
一种新型聚酮聚合催化剂的配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚的制备方法,工艺路线如图1所示;其具体步骤为:
[0073]
(1)1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚的制备:
[0074]
无水无氧条件下,将双(2-甲氧基苯基)氧化膦1.96g加入30ml四氢呋喃中,搅拌至溶解,搅拌速度为250r/min,降温至-40℃,开始控温滴加7.00ml 1.6m/ln-buli的溶液,20min后滴加完毕,缓慢升温至30℃搅拌1.5h后,降温至-10℃,开始控温滴加1,1
’‑
二溴二甲醚0.61g,20min后滴加完毕,保温反应1h后,升温至30℃,保温反应17h后,跟踪检测反应完毕,在搅拌下将反应液缓慢滴入30ml水中,滴加时间为25min,充分搅拌后,分液,水洗,干燥,减压除去溶剂后,乙醇和石油醚重结晶,烘干后得到白色的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚1.61g,纯度98.2%,收率93.44%。
[0075]
(2)1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚:
[0076]
无水无氧条件下,将1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚2.60g和三乙胺4.07g加入20ml乙腈中,控温20℃,滴加hsicl
3 5.49g,15min滴加完毕后,升温至回流状态,反应6h后,跟踪检测反应完全,冷却,加8倍hsicl3摩尔当量30wt%氢氧化钠水溶液淬灭反应,静置分液,分出有机相,乙腈萃取两次,合并有机相,饱和食盐水水洗一次,干燥,减压除去溶剂,甲醇重结晶,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚2.23g,纯度98.6%,收率91.62%。
[0077]
实施例3
[0078]
一种新型聚酮聚合催化剂的配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚的制备方法,工艺路线如图1所示;其具体步骤为:
[0079]
(1)1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚的制备:
[0080]
无水无氧条件下,将双(2-甲氧基苯基)氧化膦2.35g加入20ml四氢呋喃中,搅拌至溶解,搅拌速度为200r/min,降温至-10℃,分5批次缓慢加入叔丁醇钠3.44g,15min后加完后缓慢升温至30℃搅拌3h后,降温至-20℃,开始控温滴加1,1
’‑
二溴二甲醚0.61g,20min后滴加完毕,保温反应1h后,升温至30℃,保温反应19h后,跟踪检测反应完毕后,在搅拌下将反应液缓慢滴入30ml水中,滴加时间为25min,充分搅拌后,分液,水洗,干燥,减压除去溶剂后,乙醇和石油醚重结晶,烘干后得到白色的1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚1.56g,纯度98.1%,收率90.45%。
[0081]
(2)1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚:
[0082]
无水无氧条件下,将1,1
’‑
双[双(2-甲氧基苯基)膦酰基]二甲醚2.60g和三乙胺2.72g加入30ml乙腈中,控温30℃,分3批次缓慢加入四氢铝锂1.54g,10min后加完后升温至回流状态,反应10h后,跟踪检测反应完全,冷却,加8倍四氢铝锂摩尔当量30wt%氢氧化钠水溶液淬灭反应,静置分液,分出有机相,乙腈萃取两次,合并有机相,饱和食盐水水洗一次,干燥,减压除去溶剂,甲醇重结晶,得到1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚2.16g,
纯度98.0%,收率88.2%。
[0083]
通过以上实施例,可得到一种新型聚酮聚合催化剂的配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚,具体结构如下:
[0084][0085]
谱图如图2~图3所示。
[0086]
应用实施例
[0087]
以配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚开展聚合反应,具体步骤如下:
[0088]
在500ml高压反应釜中加入250ml甲醇,9.7mg对苯醌,占甲醇质量1%的三氟甲磺酸,1.68mg醋酸钯,4.01mg1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚;加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯15g,充入co和c2h4质量比为1:1.1的混合气至5mpa,开始升温,设定温度85℃,搅拌速度400r/min,当温度升至85℃时,持续补入co和c2h4质量比为1:1.1的混合气,维持反应压力为5.0mpa,反应时间6h;经处理干燥的产品质量121g,催化活性为25.37kg/(g-pd
·
h)。
[0089]
对比例1
[0090]
以配体3,3-双-[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂-螺[5,5]十一烷开展聚合反应,具体步骤如下:
[0091]
在500ml高压反应釜中加入250ml甲醇,9.7mg对苯醌,占甲醇质量1%的三氟甲磺酸,1.68mg醋酸钯,5.04mg3,3-双-[双-(2-甲氧基苯基)膦甲基]-1,5-二氧杂-螺[5,5]十一烷;加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯15g,充入co和c2h4质量比为1:1.1的混合气至5mpa,开始升温,设定温度85℃,搅拌速度400r/min,当温度升至85℃时,持续补入co和c2h4质量比为1:1.1的混合气,维持反应压力为5.0mpa,反应时间6h;经处理干燥的产品质量104g,催化活性为21.80kg/(g-pd
·
h)。
[0092]
对比例2
[0093]
以配体1,3-双[二(2-甲氧基苯基)膦基]丙烷(bdompp)开展聚合反应,具体步骤如下:
[0094]
在500ml高压反应釜中加入250ml甲醇,9.7mg对苯醌,占甲醇质量1%的三氟甲磺酸,1.68mg醋酸钯,3.99mg1,3-双[二(2-甲氧基苯基)膦基]丙烷;加入上述物质后,高压釜内充入氮气保压、置换,然后充入丙烯15g,充入co和c2h4质量比为1:1.1的混合气至5mpa,开始升温,设定温度85℃,搅拌速度400r/min,当温度升至85℃时,持续补入co和c2h4质量比为1:1.1的混合气,维持反应压力为5.0mpa,反应时间6h;经处理干燥的产品质量86g,催化活性为18.03kg/(g-pd
·
h)。
[0095]
实验结果表明,在相同聚合条件下,本发明提供的配体1,1
’‑
双[双(2-甲氧基苯基)膦基]二甲醚的催化活性为25kg/(g-pd
·
h),高于现有已知配体制备催化剂的催化活性。
[0096]
所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对
这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。