c7)烷基,卤素,no2和conh2;
[0018]-v选自2至4;
[0019]-r3选自直链或支链(c
1-c7)烷基,(c
1-c7)烷基-co2z和直链或支链(c
1-c7)烷基-ny1y2;所述直链或支链(c
1-c7)烷基-ny1y2任选地被(c
1-c7)烷基-co2z取代;
[0020]-r4选自:h,直链或支链(c
2-c7)烷基,直链或支链(c
2-c7)烯基,-conr7r8,芳基,杂芳基,(c
2-c7)环烷基,直链或支链-(c
1-c7)烷基-芳基和直链或支链-(c
1-c7)烷基-杂芳基;
[0021]
所述芳基,(c
2-c7)环烷基和杂芳基任选地被一个或多个选自以下的取代基取代:卤素,任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷基,任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷氧基,-cooh,芳基,-nrr’,-no2,或所述芳基和杂芳基任选地稠合以形成杂环烷基;
[0022]-r5和r6相同或不同,独立地选自:
[0023]
·
h和直链或支链(c
1-c7)烷基,或
[0024]
·
r5和r6连接在一起以与它们所连接的碳原子形成环烷基,芳基或杂芳基,或
[0025]
·
r5为h且r1和r6连接在一起以与连接至r1的氮原子形成杂环烷基或杂芳基,或
[0026]
·
r6为h且r1和r5一起连接至r1以与连接至r1的氮原子形成杂环烷基;
[0027]-r7为-(c
1-c3)烷基;
[0028]-r8为-(c
1-c3)烷基nrr’;
[0029]-r和r’相同或不同,独立地选自h和直链或支链(c
1-c7)烷基,
[0030]-y1和y2相同或不同,独立地选自h和-co-(c
1-c7)烷基;
[0031]-z选自h和直链或支链(c
1-c7)烷基;
[0032]
并且其中当x为s且r3为直链或支链(c
1-c7)烷基时,r1和r2中的至少一个为chr5chr6or4或(chr5)vor4;
[0033]
或其药学上可接受的盐或旋光异构体,外消旋体,非对映异构体,对映异构体或互变异构体。
[0034]
在一个实施方案中,根据本发明的化合物是如上所述的式(i)的化合物,其中:
[0035]-x是选自o或s的原子;
[0036]-r1和r2相同或不同,独立地选自:直链或支链(c
1-c7)烷基,直链或支链(c
2-c7)烯基,芳基,杂芳基,chr5chr6or4和(chr5)vor4,
[0037]
所述芳基和杂芳基任选地被一个或多个选自以下的取代基取代:直链或支链(c
1-c7)烷基,卤素,no2和conh2;
[0038]-v选自2至4;
[0039]-r3选自直链或支链(c
1-c7)烷基和直链或支链(c
1-c7)烷基-ny1y2;所述直链或支链(c
1-c7)烷基-ny1y2任选地被(c
1-c7)烷基-co2z取代;
[0040]-r4选自:h,直链或支链(c
2-c7)烷基,直链或支链(c
2-c7)烯基,芳基,杂芳基,直链或支链-(c
1-c7)烷基-芳基和直链或支链-(c
1-c7)烷基-杂芳基;
[0041]
所述芳基和杂芳基任选地被一个或多个选自以下的取代基取代:任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷基,任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷氧基,-cooh,芳基,-nrr’,-no2,或所述芳基和杂芳基任选地稠合以形成杂环烷基;
[0042]-r5和r6相同或不同,独立地选自:
[0043]
·
h和直链或支链(c
1-c7)烷基,或
[0044]
·
r5和r6连接在一起以与它们所连接的碳原子形成环烷基,芳基或杂芳基,或
[0045]
·
r5为h且r1和r6连接在一起以与连接至r1的氮原子形成杂环烷基或杂芳基,或
[0046]
·
r6为h且r1和r5一起连接至r1以与连接至r1的氮原子形成杂环烷基;
[0047]-r和r’相同或不同,独立地选自h和直链或支链(c
1-c7)烷基,
[0048]-y1和y2相同或不同,独立地选自h和-co-(c
1-c7)烷基;
[0049]-z选自h和直链或支链(c
1-c7)烷基;
[0050]
并且其中当x为s且r3为直链或支链(c
1-c7)烷基时,r1和r2中的至少一个为chr5chr6or4或(chr5)vor4;
[0051]
或其药学上可接受的盐或旋光异构体,外消旋体,非对映异构体,对映异构体或互变异构体。
[0052]
在另一个实施方案中,根据本发明的化合物是如上所述的式(i)的化合物,其中x为o且r3选自乙基或甲基。
[0053]
根据另一个实施方案,根据本发明的化合物是如上所述的式(i)的化合物,其中x为s,r3为直链或支链(c
1-c7)烷基,优选甲基,r1为直链或支链(c
1-c7)烷基,优选甲基,r2为chr5chr6or4或(chr5)vor4,且r5和r6为:
[0054]-h,或
[0055]-r5为h且r1和r6连接在一起以与连接至r1的氮原子形成杂环烷基,优选吡咯烷基,或
[0056]-r6为h且r1和r5一起连接至r1以与连接至r1的氮原子形成杂环烷基,优选吡咯烷基。
[0057]
根据另一个实施方案,根据本发明的化合物是如上所述的式(i)的化合物,其中x为s,r3为直链或支链(c
1-c7)烷基,r1为直链或支链(c
1-c7)烷基,且r2为chr5chr6or4或(chr5)vor4,特别是chr5chr6or4。
[0058]
特别地,r4选自h,直链或支链(c
2-c7)烷基,直链或支链(c
2-c7)烯基,-conr7r8,(c
2-c7)环烷基,直链或支链-(c
1-c7)烷基-杂芳基,芳基或苄基;所述(c
2-c7)环烷基被一个或多个选自以下的取代基取代:直链或支链(c1-c7)烷基;所述苄基任选地被一个或多个选自以下的取代基取代:任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷基,任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷氧基,卤素,或所述苄基任选地稠合以形成1,3-苯并二氧杂环戊二烯(1,3-benzodioxole)。
[0059]
可供选择地,特别地,r4选自h,直链或支链(c
2-c7)烷基,直链或支链(c
2-c7)烯基,直链或支链-(c
1-c7)烷基-杂芳基,芳基,直链或支链-(c
1-c7)烷基-芳基或苄基;所述苄基任选地被一个或多个选自以下的取代基取代:任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷基,任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷氧基,卤素或吡啶基,或所述苄基任选地稠合以形成1,3-苯并二氧杂环戊二烯。
[0060]
更特别地,r5和r6为h,且r4选自h,直链或支链(c
2-c7)烷基,直链或支链(c
2-c7)烯基,conr7r8,(c
2-c7)环烷基,直链或支链-(c
1-c7)烷基-杂芳基或苄基;所述(c
2-c7)环烷基被一个或多个选自以下的取代基取代:直链或支链(c1-c7)烷基;所述苄基任选地被一个或多
个选自以下的取代基取代:任选地被一个或多个卤素原子取代的直链或支链(c1-c7)烷基,任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷氧基,卤素。
[0061]
可供选择地,更特别地,r5和r6为h,且r4选自h,直链或支链(c
2-c7)烷基,直链或支链(c
2-c7)烯基,直链或支链-(c
1-c7)烷基-杂芳基,直链或支链-(c
1-c7)烷基-芳基或苄基;所述苄基任选地被一个或多个选自以下的取代基取代:任选地被一个或多个卤素原子取代的直链或支链(c1-c7)烷基,任选地被一个或多个卤素原子取代的直链或支链(c
1-c7)烷氧基,卤素。
[0062]
甚至更特别地,r1为甲基,且r4选自:h,conr7r8,其中r7为甲基且r8为nrr’,其中r和r’为甲基,乙基,丙烯,苄基,吡啶基,苄基氧基丁基,被一个或多个甲基取代的甲基-环己烯基,和被一个或多个氟,氯,甲氧基或甲基取代的苄基。
[0063]
可供选择地,甚至更特别地,r1为甲基,且r4选自:h,乙基,丙烯,苄基,吡啶基,苄基氧基丁基和被一个或多个氟,氯,甲氧基或甲基取代的苄基。
[0064]
在另一个实施方案中,x为s,r1和r2为直链或支链(c
1-c7)烷基,且r3为-(c
1-c7)-co2z或直链或支链(c
1-c7)烷基-ny1y2,所述直链或支链(c
1-c7)烷基-ny1y2任选地被(c
1-c7)-co2z取代,特别地,x为s,r1和r2为直链或支链(c
1-c7)烷基,且r3为直链或支链(c
1-c7)烷基-ny1y2,所述直链或支链(c
1-c7)烷基-ny1y2任选地被(c
1-c7)-co2z取代。
[0065]
特别地,y1和y2相同或不同,独立地选自h和-co-ch3。
[0066]
更特别地,z选自h和叔丁基(第三丁基)。
[0067]
仍然特别地,r3为直链或支链(c
1-c3)烷基-ny1y2。
[0068]
根据具体的实施方案,式(i)的化合物选自:
[0069]-4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0070]-4-[2-烯丙基氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0071]-4-[2-苄基氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0072]-4-甲基-4-[甲基-[2-(间甲苯基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯;
[0073]-4-[2-[(3,4-二甲基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0074]-4-[2-[(4-甲氧基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0075]-4-[2-[(3,4-二甲氧基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0076]-4-[2-[(3-氯苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0077]-4-[2-[(3-氟苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0078]-4-甲基-4-[甲基-[2-(2-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯;
[0079]-4-甲基-4-[甲基-[2-(3-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯;
[0080]-4-甲基-4-[甲基-[2-(4-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯;
[0081]-4-(二甲基氨基)-4-甲基-戊-2-炔酸甲酯;
[0082]-4-(二甲基氨基)-4-甲基-戊-2-炔酸乙酯;
[0083]-2-氨基-3-((4-(二甲基氨基)-4-甲基戊-2-炔酰基)硫基)丙酸;
[0084]-2-氨基-4-((4-二甲基氨基)-4-甲基戊基-2-炔酰基)硫基)丁酸;
[0085]-2-乙酰氨基-3-((4-(二甲基氨基)-4-甲基戊-2-炔酰基)硫基)丙酸乙酯;
[0086]-2-((4-(二甲基氨基)-4-甲基戊-2-炔酰基)硫基)乙酸叔丁酯;
[0087]-2-((4-(二甲基氨基)-4-甲基戊-2-炔酰基)硫基)乙酸;
[0088]-4-((4-(苄基氧基)丁基)(甲基)氨基)-4-甲基戊-2-炔硫代酸s-甲酯;
[0089]-4-((2-羟基乙基)(甲基)氨基)-4-甲基戊-2-炔硫代酸s-甲酯;
[0090]-4-甲基-4-[甲基-[2-(2-萘基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯;
[0091]-4-甲基-4-[甲基-[2-[(2,6,6-三甲基环己烯-1-基)甲氧基]乙基]氨基]戊-2-炔硫代酸s-甲酯;
[0092]-3,4-二甲氧基苯甲酸2-[(1,1-二甲基-4-甲基硫烷基-4-氧代-丁-2-炔基)-甲基氨基]乙酯;
[0093]-乙酸2[(1,1-二甲基-4-甲基硫烷基-4-氧代-丁-2-炔基)-甲基氨基]乙酯;
[0094]-2,5,10,11,11-五甲基-6-氧代-7-氧杂-2,5,10-三氮杂十四碳-12-炔-14-硫代酸s-甲酯;
[0095]-4-[2-(甲氧基甲基)吡咯烷-1-基]-4-甲基戊-2-炔硫代酸s-甲酯;
[0096]-4-(3-甲氧基吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯;
[0097]-4-甲基-4-[甲基(2-苯氧基环戊基)氨基]戊-2-炔硫代酸s-甲酯;
[0098]-(s)-4-(2-((苄基氧基)甲基)吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯;
[0099]-4-[(3(苄基氧基)-1吡咯烷基])-4-甲基戊-2-炔硫代酸s-甲酯,
[0100]
或其药学上可接受的盐或旋光异构体,外消旋体,非对映异构体,对映异构体或互变异构体。
[0101]
根据具体的实施方案,式(i)的化合物选自:
[0102]-4-[2-苄基氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;和
[0103]-4-[2-[(3,4-二甲基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯;
[0104]
或其药学上可接受的盐或旋光异构体,外消旋体,非对映异构体,对映异构体或互变异构体。
[0105]
在另一个实施方案中,根据本发明的化合物是如上所述的式(i)的化合物,其中:
[0106]-x为s;
[0107]-r1为直链或支链(c
1-c7)烷基;
[0108]-r2为chr5chr6or4或(chr5)vor4;
[0109]-r4选自h,芳基,杂芳基,直链或支链-(c
1-c7)烷基-芳基和直链或支链-(c
1-c7)烷基-杂芳基;
[0110]
所述芳基和杂芳基任选地被一个或多个选自以下的取代基取代:-cooh,-nrr’和-no2;并且
[0111]-r和r’相同,为h。
[0112]
本发明还涉及制备如本文所述的式(i)的化合物的方法,所述方法包括:
[0113]
a)使式(ii)的化合物与有机酸或无机酸反应
[0114][0115]
b)使步骤a)中获得的化合物与碱反应;
[0116]
c)使步骤b)中获得的化合物与co2反应;
[0117]
d)使步骤c)中获得的化合物与氯甲酸烷基酯,能够与步骤c)中获得的化合物形成酰基卤的试剂或能够与步骤c)中获得的化合物形成混合酸酐的试剂反应;
[0118]
e)使步骤d)中获得的化合物与阴离子前体化合物sme-反应;
[0119]
其中r1和r2如本文所定义。
[0120]
特别地,本发明涉及如上所述的方法,其中步骤b)的碱具有大于25的pka,优选地,步骤b)中使用的碱选自锂碱或镁碱,优选地,所述碱选自丁基锂或己基锂。
[0121]
本发明还涉及药物组合物,其包含如本文所述的式(i)的化合物和药学上可接受的赋形剂。
[0122]
本发明还涉及如本文所述的式(i)的化合物,其用作药物。
[0123]
特别地,本发明涉及如本文所述的式(i)的化合物,其用于预防或治疗癌症。
[0124]
更特别地,本发明涉及如本文所述的式(i)的化合物,其用于预防或治疗白血病。
[0125]
本发明还涉及式b-l-ab的抗体药物缀合物,其中:
[0126]-b为如上所述的式(i)的化合物,其中:
[0127]-x为s;
[0128]-r1为直链或支链(c
1-c7)烷基;
[0129]-r2为chr5chr6or4或(chr5)vor4;
[0130]-r4选自h,芳基,杂芳基,直链或支链-(c
1-c7)烷基-芳基和直链或支链-(c
1-c7)烷基-杂芳基;
[0131]
所述芳基和杂芳基任选地被一个或多个选自以下的取代基取代:-cooh,-nrr’和-no2;并且
[0132]-r和r’相同,为h;
[0133]-l为接头;并且
[0134]-ab为抗体。
[0135]
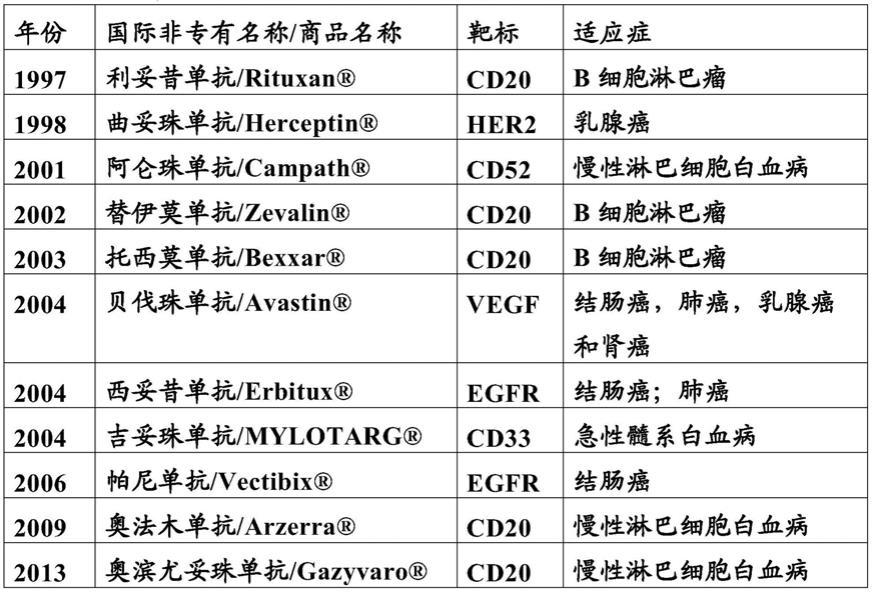
特别地,所述抗体药物缀合物的抗体选自:利妥昔单抗,曲妥珠单抗,阿仑珠单抗,替伊莫单抗(ibritumomab),替伊莫单抗(tiuxetan),托西莫单抗,贝伐珠单抗(brevacizumab),西妥昔单抗,帕尼单抗,奥法木单抗,伊匹木单抗和奥滨尤妥珠单抗(obinutuzumab)。
[0136]
根据本发明的式(i)的化合物如上所述。
[0137]
本发明还涉及任何以下实施方案或其任何组合,条件是当r3为直链或支链(c
1-c7)烷基且x为s时,r1和r2中的至少一个为chr5chr6or4。
[0138]
在一个实施方案中,x为s。
[0139]
在另一个实施方案中,r3为甲基。
元单环,双环或多环,其可以任选地被桥接,并且其中环的至少一个成员是杂原子。通常,杂原子包括但不限于o或n。特别地,每个环包含1至3个杂原子。合适的杂环也在handbook of chemistry and physics,第76版,crc press,inc.,1995-1996,第225-226页中公开,其公开内容通过引用并入本文。杂环烷基的实例包括但不限于四氢吡啶基,四氢吡喃基,吡咯烷基,哌啶基,吗啉基,咪唑烷基或苯并二氧杂环戊二烯,特别是1,3-苯并二氧杂环戊二烯。
[0153]-除非另有说明,术语“取代的”是指用一个或多个取代基取代,这些取代基可以相同或不同,例如选自直链或支链(c
1-c7)烷基,卤素,no2和conh2,被一个或多个卤素原子取代的直链或支链(c
1-c7)烷基,直链或支链(c
1-c7)烷氧基,被一个或多个卤素原子取代的直链或支链(c
1-c7)烷氧基,芳基,-cooh,-cooch2ch3,-nrr’,nh2,nh烷基和n(烷基)2。实例特别地包括甲基,甲氧基,氯,氟,cf3和ocf3。
[0154]
如本文所述的式(i)的化合物可以包含一个或多个不对称碳原子。因此它们可以以对映异构体或非对映异构体的形式存在。这些对映异构体和非对映异构体以及它们的混合物,包括外消旋混合物,构成本发明的一部分。
[0155]
本文所述的式(i)的化合物可以游离碱形式或以与酸的加成盐的形式提供,这也构成本发明的一部分。
[0156]
这些盐有利地用药学上可接受的酸制备,但与其他酸形成的盐,例如用于纯化或分离如本文所述的式(i)的化合物的盐,也构成本发明的一部分。
[0157]
如本文所使用,表述“药学上可接受的”是指在合理的医学判断范围内,适合与人类和动物的组织接触而没有过度毒性,刺激性,过敏反应或其他有问题的并发症的与合理的收益/风险比相称的那些化合物,材料,赋形剂,组合物或剂型。
[0158]
如本文所使用,“药学上可接受的盐”是指所公开的化合物的衍生物,其中母体化合物通过制备其酸或碱盐而被修饰。药学上可接受的盐包括例如由无毒无机或有机酸形成的母体化合物的常规无毒盐或季铵盐。例如,这样的常规无毒盐包括:衍生自诸如盐酸,氢溴酸,硫酸,氨基磺酸,磷酸,硝酸等的无机酸的那些,包括其单盐,二盐或三盐;以及由诸如乙酸,丙酸,琥珀酸,酒石酸,柠檬酸,甲磺酸,苯磺酸,葡萄糖醛酸,谷氨酸,苯甲酸,水杨酸,甲苯磺酸,草酸,富马酸,马来酸,乳酸等的有机酸制备的盐。另外的加成盐包括诸如氨丁三醇,葡甲胺,吡咯乙醇等的铵盐,诸如钠,钾,钙,锌或镁的金属盐。
[0159]
本发明的药学上可接受的盐可以由含有碱性或酸性部分的母体化合物通过常规化学方法合成。通常,这样的盐可以通过使这些化合物的游离酸或碱形式与化学计量的量的适宜的碱或酸在水中或有机溶剂中或在两者的混合物中反应来制备。通常,优选非水性介质,如醚,乙酸乙酯,乙醇,异丙醇或乙腈。合适的盐的列表见remington’s pharmaceutical sciences,第20版,mack publishing company,easton,pa,2000,其公开内容通过引用并入本文。
[0160]
方法
[0161]
本发明还涉及如本文所述的式(i)的化合物的制备方法。
[0162]
本发明的化合物和方法可以以本领域技术人员熟知的多种方式制备。化合物可以例如通过应用或调整下述方法或如本领域技术人员所理解的其变化来合成。对本领域技术人员而言,适当的修改和替换将是显而易见的和众所周知的或容易从科学文献中获得。
[0163]
应当理解,本发明的化合物可以含有一个或多个不对称取代的碳原子,并且可以
以旋光或外消旋形式分离。因此,结构的所有手性,非对映异构,外消旋形式,异构形式都是预期的,除非具体指出具体的立体化学或异构形式。如何制备和分离这样的旋光形式在本领域中是众所周知的。
[0164]
例如,立体异构体的混合物可以通过标准技术(包括但不限于外消旋形式的拆分,正相,反相和手性色谱,优先成盐,重结晶等)来分离,或通过手性合成由手性起始材料或通过目标手性中心的刻意合成来分离。
[0165]
本发明的化合物可以通过多种合成途径制备。试剂和起始材料是可商购的,或由本领域普通技术人员通过众所周知的技术容易地合成。除非另有说明,所有取代基均如前所定义。
[0166]
在下文描述的反应中,可能需要保护在终产物中期望的反应性官能团,例如羟基,氨基,亚氨基,硫醇基(thio)或羧基,以避免它们不希望地参与反应。可以根据标准实践使用常规保护基团,例如参见t.w.greene和p.g.m.wuts的protective groups in organic chemistry,第4版(2007),john wiley&sons inc.,1999;j.f.w.mcomie的protective groups in organic chemistry,plenum press,1973。
[0167]
一些反应可以在碱的存在下进行。对用于该反应的碱的性质没有特别限制,任何常规用于此类反应的碱都可以在此同样使用,只要其对分子的其他部分没有不利影响以及除非另有说明。合适的碱的实例包括:氢氧化钠,碳酸钾,三乙胺,碱金属氢化物,例如氢化钠和氢化钾;烷基锂化合物,例如甲基锂和丁基锂;和碱金属醇盐,例如甲醇钠和乙醇钠。
[0168]
通常,反应在合适的溶剂中进行。可以使用多种溶剂,只要其对反应或所涉及的试剂没有不利影响。合适的溶剂的实例包括:烃,其可以是芳族烃,脂族烃或脂环族烃,例如己烷,环己烷,苯,甲苯和二甲苯;酰胺,例如二甲基甲酰胺;醇,例如乙醇和甲醇;以及醚,例如乙醚和四氢呋喃。
[0169]
反应可以在宽的温度范围内发生。通常,发现在0℃至150℃(更优选约室温至100℃)的温度下进行反应是便利的。反应所需的时间也可以有很大差异,这取决于许多因素,特别是反应温度和试剂的性质。然而,只要反应在上述优选条件下进行,3小时至20小时的时间段通常就足够了。
[0170]
可以通过常规方法从反应混合物中回收如此制备的化合物。例如,可以通过从反应混合物中蒸馏出溶剂来回收化合物,或者,如果需要,在从反应混合物中蒸馏出溶剂之后,将残余物倒入水中,然后用与水不混溶的有机溶剂萃取并从提取物中蒸馏出溶剂。此外,如果需要,产物可以通过各种众所周知的技术进一步纯化,例如重结晶,再沉淀或各种色谱技术,特别是柱色谱或制备薄层色谱。
[0171]
本发明的式(i)的化合物的制备方法是本发明的另一个目的。
[0172]
根据第一方面,本发明的式(i)的化合物可以通过以下步骤获得:
[0173]
a)使式(ii)的化合物与有机酸或无机酸反应
[0174]
[0175]
b)使步骤a)中获得的化合物与碱反应;
[0176]
c)使步骤b)中获得的化合物与co2反应;
[0177]
d)使步骤c)中获得的化合物与氯甲酸烷基酯,能够与步骤c)中获得的化合物形成酰基卤的试剂或能够与步骤c)中获得的化合物形成混合酸酐的试剂反应;
[0178]
e)使步骤d)中获得的化合物与阴离子前体化合物sme-反应;
[0179]
其中r1和r2如本文所定义。
[0180]
特别地,步骤b)的碱具有大于25的pka,优选地,步骤b)中使用的碱选自锂碱或镁碱,优选地,所述碱选自丁基锂或己基锂。
[0181]
特别地,通过步骤a1)的3-氯-3-甲基丁-1-炔与r1r2nh在水性介质中的反应来获得式(ii)的化合物。
[0182]
特别地,步骤a1)中获得的所述化合物通过一次或多次过滤纯化,例如过滤或连续2至10次过滤,优选连续2至5次过滤,例如4次过滤。
[0183]
在一个实施方案中,3-氯-3-甲基丁-1-炔通过在铜催化剂存在下使2-甲基丁-3-炔-2-醇与盐酸反应的反应步骤来获得。
[0184]
在另一个实施方案中,酸是选自盐酸,磷酸,硝酸,硫酸的无机酸,优选盐酸。
[0185]
在另一个实施方案中,步骤d)用以下进行:
[0186]-具有(c
1-c6)烷基的氯甲酸烷基酯,其可以包含至少一个双键,优选氯甲酸甲酯,氯甲酸乙酯,氯甲酸异戊二烯酯,氯甲酸叔丁酯或氯甲酸异丁酯,优选氯甲酸异丁酯;或
[0187]-能够与步骤c)中获得的化合物形成混合酸酐的试剂,其选自酰氯,例如新戊酰氯;或
[0188]-能够与步骤c)中获得的化合物形成酰基卤的试剂,其选自socl2,cocl2,pcl3,pcl5,pbr3或pph
3 br2。
[0189]
在一个实施方案中,阴离子前体化合物sme-选自式xsme的盐,其中x表示碱金属或碱土金属,例如na,甲硫醇或(sme)2,优选nasme。
[0190]
该方法在专利申请fr1651283和pct/ep2017/053457中有详细描述,其内容通过引用并入本文。
[0191]
可供选择地,根据本发明的化合物可以由相应的炔胺依次用buli,cos和mei处理来制备。详细的制备方法可以见于例如g.quash等人,european journal of medicinal chemistry 43(2008)906-916,其内容通过引用并入本文,特别是材料的和方法章节的第2部分。
[0192]
上述反应可以由技术人员通过应用或调整下文实施例中示出的方法来进行。
[0193]
此外,本发明的方法还可以包括分离式(i)或式(ii)的化合物的另外的步骤。这可以由技术人员通过任何已知的常规手段,例如上述回收方法来完成。
[0194]
通常,起始产品主要可从aldrich或acros或其他典型化学品供应商商购获得,或者可以通过应用或调整任何已知方法或实施例中描述的方法获得。
[0195]
用途
[0196]
如已经提到的,本发明还涉及如本文所述的式(i)的化合物,其用作药物。
[0197]
更特别地,本发明涉及如本文所述的式(i)的化合物,其用于预防和/或治疗癌症。
[0198]
本发明还涉及预防和/或治疗癌症的方法,所述方法包括给予有需要的受试者有
效量的如本文所述的式(i)的化合物。
[0199]
如在本发明的上下文中使用的,术语“治疗(treat)”,“治疗(treating)”,“治疗(treated)”或“治疗(treatment)”是指治疗性治疗,其中目的是消除或减轻症状。有益的或期望的临床结果包括但不限于症状的消除,症状的减轻,病症程度的减小,病症的稳定(即,不恶化)状态,病症进展的延迟或减慢。
[0200]
如在本发明的上下文中使用的,术语“预防(prevent)”,“预防(prevention)”,“预防(preventing)”或“预防(prevented)”是指预防疾病或障碍或其一种或多种症状的发作,复发或扩散。在某些实施方案中,该术语是指在症状发作之前用本文提供的化合物进行治疗,或给予本文提供的化合物,特别是给予具有本文提供的疾病或障碍的风险的患者。该术语包括抑制或减轻特定疾病的症状。在某些实施方案中,具有疾病家族史的受试者尤其是预防方案的候选者。此外,有反复出现的症状史的受试者也是预防的潜在候选人。在这方面,术语“预防”可以与术语“预防性治疗”互换使用。
[0201]
如本文所使用且除非另有定义,“癌症”是指体内异常细胞的生长,分裂或增殖。它是指任何类型的恶性(即非良性)肿瘤。恶性肿瘤可以相当于原发性肿瘤或继发性肿瘤(即转移瘤)。此外,该肿瘤可以相当于实体恶性肿瘤,其包括例如癌,腺癌,肉瘤,黑色素瘤,间皮瘤,母细胞瘤或血癌,如白血病,淋巴瘤和骨髓瘤。癌症可以例如相当于实体癌,黑色素瘤,肺癌(包括但不限于非小细胞肺癌(nsclc),小细胞肺癌(sclc),复合性小细胞癌,胸膜肺母细胞瘤,类癌瘤,肉瘤样癌,类癌瘤,腺鳞癌,鳞状细胞肺癌,腺癌和大细胞肺癌),脑癌(包括但不限于胶质瘤,胶质母细胞瘤,星形细胞瘤,少突星形细胞瘤,少突胶质瘤和室管膜瘤),肾癌,前列腺癌,乳腺癌,骨髓增生异常综合征和白血病。
[0202]
特别地,本发明涉及如本文所述的式(i)的化合物,其用于预防和/或治疗白血病。
[0203]
特别地,需要针对癌症进行治疗的受试者是患有这样的疾病的受试者。
[0204]
在本发明的上下文中,需要治疗本文所述疾病和病症的受试者的确定如上所述进行,并且完全在本领域技术人员的能力和知识范围内。本领域的临床医生可以通过上述技术容易地确定需要这样的治疗的那些受试者。
[0205]
治疗有效量可以由和本领域技术人员一样的主治诊断医生通过使用常规技术并通过观察在类似情况下获得的结果容易地确定。在确定治疗有效量时,主治诊断医生考虑了许多因素,包括但不限于:受试者的物种;其大小,年龄和一般健康状况;涉及的具体疾病;疾病的受累程度或严重程度;个体受试者的反应;给予的具体化合物;给药方式;给予的制剂的生物利用度特征;选择的剂量方案;伴随药物的使用;及其他相关情况。
[0206]
如本文所使用,“有效量”是指对减少,消除,治疗或控制本文所述疾病和病症的症状方面有效的量。术语“控制”意图指其中可能存在本文所述的疾病和病症进展的减慢,中断,阻止或停止的所有过程,但不一定表示所有疾病和病症症状的完全消除,并且意图包括预防性治疗和长期使用。
[0207]
除非另有说明,否则术语“患者”或“受试者”是指患有或可能患上一种或多种本文所述的疾病和病症的温血动物,例如哺乳动物,特别是人类男性或女性。
[0208]
实现期望生物效果所需的根据本发明的化合物的量将根据许多因素而变化,包括待给药的药物的剂量,所用化合物的化学特性(例如疏水性),化合物的效力,疾病的类型,患者的疾病状态和给药途径。
[0209]
本文提供的化合物可以配制成药物组合物,任选地通过与一种或多种药学上可接受的赋形剂混合来配制。
[0210]
这样的组合物可以制备用于口服给药,特别是以片剂或胶囊剂,特别是口分散(lyoc)片剂的形式;或肠胃外给药,特别是以液体溶液剂,混悬剂或乳剂的形式。
[0211]
其可以通过制药领域众所周知的任何方法制备,例如,如remington:the science and practice of pharmacy,第20版;gennaro,a.r.,ed.;lippincott williams&wilkins:philadelphia,pa,2000中所述。药学相容的粘合剂和/或辅助剂材料可以作为组合物的一部分包含在内。口服组合物通常包含惰性稀释剂载体或可食用载体。它们可以以单位剂量形式给药,其中术语“单位剂量”是指能够给予患者并且可以容易地处理和包装的单次剂量,其保持为包含活性化合物本身或作为药学上可接受的组合物的物理和化学稳定的单位剂量。
[0212]
片剂,丸剂,散剂,胶囊剂,锭剂等可以含有任何以下成分或具有类似性质的化合物中的一种或多种:粘合剂,例如微晶纤维素或黄蓍胶;稀释剂,例如淀粉或乳糖;崩解剂,例如淀粉和纤维素衍生物;润滑剂,例如硬脂酸镁;助流剂,例如胶体二氧化硅;甜味剂,例如蔗糖或糖精;或调味剂,例如薄荷或水杨酸甲酯。胶囊可以是通常由任选地与增塑剂掺合的明胶掺合物制成的硬胶囊或软胶囊的形式,以及淀粉胶囊的形式。此外,剂量单位形式可以包含改变剂量单位的物理形式的各种其他材料,例如糖,虫胶或肠溶剂的包衣。其他口服剂型糖浆剂或酏剂可以含有甜味剂,防腐剂,染料,色素和调味剂。此外,活性化合物可以掺入速溶,调释(modified-release)或缓释制剂(preparation)和制剂(formulation)中,其中这样的缓释制剂优选为双峰的。
[0213]
用于给药的液体制剂包括无菌水性或非水性溶液剂,混悬剂和乳剂。液体组合物还可以包含粘合剂,缓冲剂,防腐剂,螯合剂,甜味剂,调味剂和着色剂等。非水性溶剂包括醇,丙二醇,聚乙二醇,丙烯酸酯共聚物,植物油如橄榄油和有机酯如油酸乙酯。水性载体包括醇和水的混合物,水凝胶,缓冲介质和盐水。特别地,生物相容的,可生物降解的丙交酯聚合物,丙交酯/乙交酯共聚物或聚氧乙烯-聚氧丙烯共聚物可以是控制活性化合物释放的有用赋形剂。静脉内溶媒可以包括流体和营养补充剂,电解质补充剂,例如基于林格氏葡萄糖的那些,等等。
[0214]
给药方式的实例包括肠胃外给药,例如皮下给药,肌肉内给药,静脉内给药,皮内给药以及口服给药。
[0215]
抗体药物缀合物
[0216]
如前所述,本发明还涉及式b-l-ab的抗体药物缀合物,其中:
[0217]-b为如上所述的式(i)的化合物,其中:
[0218]-x为s;
[0219]-r1为直链或支链(c
1-c7)烷基;
[0220]-r2为chr5chr6or4或(chr5)vor4;
[0221]-r4选自h,芳基,杂芳基,直链或支链-(c
1-c7)烷基-芳基和直链或支链-(c
1-c7)烷基-杂芳基;
[0222]
所述芳基和杂芳基任选地被一个或多个选自以下的取代基取代:-cooh,-nrr’和-no2;并且
[0223]-r和r’相同,为h;
[0224]-l为接头;并且
[0225]-ab为抗体。
[0226]“抗体药物缀合物”或adc是指一类重要的高效生物药物,旨在作为治疗癌症患者的靶向疗法。与化学疗法不同,adc旨在仅靶向和杀死癌细胞并保留健康细胞。adc是由与生物活性细胞毒性(抗癌)有效载荷或药物连接的抗体组成的分子。在本发明的上下文中,细胞毒性药物是上述式(i)的化合物。该化合物通过接头与抗体连接。
[0227]
如本文所使用,“接头”是指包含共价键或原子链的化学部分,其将抗体共价连接至如上所述的式(i)的化合物。
[0228]
抗体药物缀合物的接头可以是任何能够偶联抗体和上述式(i)的化合物的接头。合适的连接基团是本领域众所周知的。特别地,接头可以是可生物降解的接头。
[0229]
更特别地,如上所述的根据本发明的化合物通过连接基团(马来酰亚胺,琥珀酰亚胺酯,酶的特定肽序列底物等)与抗体偶联,连接至可切割的接头(蛋白酶位点,肼,二硫化物)或不可切割的接头,并且具有或不具有自牺牲间隔基团(self-imolative spacer)。
[0230]
在抗体药物缀合物b-l-ab的上述定义中,接头l因此包括接头和最终连接至如本文定义的连接基团的接头。
[0231]
可切割的二肽接头如val-ala和val-cit可作为实例引用。其利用抗体-药物缀合物靶向机制,该机制涉及抗体-药物缀合物与其在目标癌细胞表面上的同源抗原的顺序结合,以及adc-抗原复合物通过内体-溶酶体途径的内化。
[0232]
在这些情况下,细胞毒性抗癌药物的细胞内释放依赖于这样的事实,即内体/溶酶体是酸性区室,可促进酸不稳定化学键(如腙)的切割。此外,如果将溶酶体特异性蛋白酶切割位点设计到接头中,例如vcmmae中的组织蛋白酶b位点,则细胞毒素将在其细胞内靶标附近释放。
[0233]
可供选择地,含有混合二硫化物的接头提供了另一种方法,通过该方法,细胞毒性有效载荷可以在细胞内释放,因为它们在细胞的还原环境中被选择性地切割,但在血流中的富氧环境中不会被选择性地切割。
[0234]
这些接头可以通过本领域技术人员熟知的方法制备。
[0235]
特别地,根据本发明的接头是在实验部分中描述的马来酰亚氨基己酰基-val-cit。
[0236]
可以在本发明的上下文中使用的接头的其他实例及其制备方法可以是马来酰亚氨基己酰基接头,巯基乙酰氨基己酰基,腙接头和与自牺牲接头对氨基苯甲醇(pab)组合的葡糖苷酸,如perez等人“antibody-drug conjugates:current status and future directions”;drug discovery today,第00卷,第00期,2013年12月以及mccombs和shawn:“antibody drug conjugates:design and selection of linker,payload and conjugation chemistry”,the aaps journal,第17卷,第2期,2015年3月中提到的。
[0237]
根据本发明的抗体药物缀合物的抗体可以是已知用于治疗癌症的任何抗体。
[0238]
特别地,所述抗体药物缀合物的抗体选自:利妥昔单抗,曲妥珠单抗,阿仑珠单抗,替伊莫单抗(ibritumomab tiuxetan),托西莫单抗,贝伐珠单抗,西妥昔单抗,帕尼单抗,奥法木单抗,伊匹木单抗和奥滨尤妥珠单抗。
[0239]
这些抗体是可商购的并且是本领域技术人员熟知的。
[0240]
下表1中给出了有关这些抗体的更多信息。
[0241][0242]
制备根据本发明的抗体药物缀合物的方法将由本领域技术人员根据所选接头和抗体的功能进行调整。本领域技术人员将能够在其一般知识的基础上制备抗体药物缀合物。实例在本发明的实验部分给出。
[0243]
本发明还涉及如上定义的抗体药物缀合物,其用作药物,特别是用于预防和/或治疗癌症。
[0244]
本发明还涉及预防和/或治疗癌症的方法,所述方法包括给予根据本发明的抗体药物缀合物。
[0245]
所治疗的癌症的实例是b细胞淋巴瘤,乳腺癌,慢性淋巴细胞白血病,结肠癌,肺癌,乳腺癌,肾癌和黑色素瘤。
[0246]
本发明还涉及包含根据本发明的抗体药物缀合物的药物组合物。
[0247]
如在本发明的上下文中使用的,术语“治疗(treat)”,“治疗(treating)”,“治疗(treated)”或“治疗(treatment)”是指治疗性治疗,其中目的是消除或减轻症状。有益的或期望的临床结果包括但不限于症状的消除,症状的减轻,病症程度的减小,病症的稳定(即,不恶化)状态,病症进展的延迟或减慢。
[0248]
如在本发明的上下文中使用的,术语“预防(prevent)”,“预防(prevention)”,“预防(preventing)”或“预防(prevented)”是指预防疾病或障碍或其一种或多种症状的发作,复发或扩散。在某些实施方案中,该术语是指在症状发作之前用本文提供的化合物进行治疗,或给予本文提供的化合物,特别是给予具有本文提供的疾病或障碍的风险的患者。该术语包括抑制或减轻特定疾病的症状。在某些实施方案中,具有疾病家族史的受试者尤其是预防方案的候选者。此外,有反复出现的症状史的受试者也是预防的潜在候选人。在这方面,术语“预防”可以与术语“预防性治疗”互换使用。
[0249]
如本文所使用且除非另有定义,“癌症”是指体内异常细胞的生长,分裂或增殖。它是指任何类型的恶性(即非良性)肿瘤。恶性肿瘤可以相当于原发性肿瘤或继发性肿瘤(即转移瘤)。
[0250]
特别地,需要针对癌症进行治疗的受试者是患有这样的疾病的受试者。
[0251]
在本发明的上下文中,需要治疗本文所述疾病和病症的受试者的确定如上所述进行,并且完全在本领域技术人员的能力和知识范围内。本领域的临床医生可以通过上述技术容易地确定需要这样的治疗的那些受试者。
[0252]
治疗有效量可以由和本领域技术人员一样的主治诊断医生通过使用常规技术并通过观察在类似情况下获得的结果容易地确定。在确定治疗有效量时,主治诊断医生考虑了许多因素,包括但不限于:受试者的物种;其大小,年龄和一般健康状况;涉及的具体疾病;疾病的受累程度或严重程度;个体受试者的反应;给予的具体化合物;给药方式;给予的制剂的生物利用度特征;选择的剂量方案;伴随药物的使用;及其他相关情况。
[0253]
如本文所使用,“有效量”是指对减少,消除,治疗或控制本文所述疾病和病症的症状方面有效的量。术语“控制”意图指其中可能存在本文所述的疾病和病症进展的减慢,中断,阻止或停止的所有过程,但不一定表示所有疾病和病症症状的完全消除,并且意图包括预防性治疗和长期使用。
[0254]
除非另有说明,否则术语“患者”或“受试者”是指患有或可能患上一种或多种本文所述的疾病和病症的温血动物,例如哺乳动物,特别是人类男性或女性。
[0255]
实现期望生物效果所需的根据本发明的抗体药物缀合物的量将根据许多因素而变化,包括待给药的剂量,所用化合物的化学和生物特性(例如疏水性),化合物的效力,疾病的类型,患者的疾病状态和给药途径。
[0256]
本文提供的抗体药物缀合物可以配制成药物组合物,任选地通过与一种或多种药学上可接受的赋形剂混合来配制。
[0257]
这样的组合物可以制备用于口服给药,特别是以片剂或胶囊剂,特别是口分散(lyoc)片剂的形式;或肠胃外给药,特别是以液体溶液剂,混悬剂或乳剂的形式。
[0258]
其可以通过制药领域众所周知的任何方法制备,例如,如remington:the science and practice of pharmacy,第20版;gennaro,a.r.,ed.;lippincott williams&wilkins:philadelphia,pa,2000中所述。药学相容的粘合剂和/或辅助剂材料可以作为组合物的一部分包含在内。口服组合物通常包含惰性稀释剂载体或可食用载体。它们可以以单位剂量形式给药,其中术语“单位剂量”是指能够给予患者并且可以容易地处理和包装的单次剂量,其保持为包含活性化合物本身或作为药学上可接受的组合物的物理和化学稳定的单位剂量。
[0259]
片剂,丸剂,散剂,胶囊剂,锭剂等可以含有任何以下成分或具有类似性质的化合物中的一种或多种:粘合剂,例如微晶纤维素或黄蓍胶;稀释剂,例如淀粉或乳糖;崩解剂,例如淀粉和纤维素衍生物;润滑剂,例如硬脂酸镁;助流剂,例如胶体二氧化硅;甜味剂,例如蔗糖或糖精;或调味剂,例如薄荷或水杨酸甲酯。胶囊可以是通常由任选地与增塑剂掺合的明胶掺合物制成的硬胶囊或软胶囊的形式,以及淀粉胶囊的形式。此外,剂量单位形式可以包含改变剂量单位的物理形式的各种其他材料,例如糖,虫胶或肠溶剂的包衣。其他口服剂型糖浆剂或酏剂可以含有甜味剂,防腐剂,染料,色素和调味剂。此外,活性化合物可以掺
入速溶,调释或缓释制剂(preparation)和制剂(formulation)中,其中这样的缓释制剂优选为双峰的。
[0260]
用于给药的液体制剂包括无菌水性或非水性溶液剂,混悬剂和乳剂。液体组合物还可以包含粘合剂,缓冲剂,防腐剂,螯合剂,甜味剂,调味剂和着色剂等。非水性溶剂包括醇,丙二醇,聚乙二醇,丙烯酸酯共聚物,植物油如橄榄油和有机酯如油酸乙酯。水性载体包括醇和水的混合物,水凝胶,缓冲介质和盐水。特别地,生物相容的,可生物降解的丙交酯聚合物,丙交酯/乙交酯共聚物或聚氧乙烯-聚氧丙烯共聚物可以是控制活性化合物释放的有用赋形剂。静脉内溶媒可以包括流体和营养补充剂,电解质补充剂,例如基于林格氏葡萄糖的那些,等等。
[0261]
给药方式的实例包括肠胃外给药,例如皮下给药,肌肉内给药,静脉内给药,皮内给药以及口服给药。
[0262]
在本发明的范围内,必须理解“用于治疗或预防
……
的化合物”等同于“化合物用于治疗或预防
……
的用途”和“化合物在制备用于治疗或预防
……
的药物中的用途”。
[0263]
本发明将通过以下附图和实施例进一步说明。
附图说明
[0264]
图1:在50μg/ml利妥昔单抗(85.50%
±
2.268%)和与化合物5偶联的利妥昔单抗(102.9%
±
1.789%)处理后,raji细胞与未处理细胞相比的平均生存力百分比。使用未配对t检验时,这两种方法之间的差异是显著的(p《0.01,**)。
实施例
[0265]
本发明的代表性化合物总结于下表2中:
[0266]
表2
[0267]
[0268]
[0269]
[0270]
[0271][0272]
本发明的代表性化合物可以根据以下程序合成。
[0273]
通用分析程序
[0274]1h和
13
c nmr光谱在来自bruker的bruker advance als300和drx400mhz上记录。化学位移以ppm(δ)为单位报告,以dmso-d6(1h,2.50ppm;
13
c,39.52ppm)或cdcl3(7.26ppm)为参考。耦合常数(j)以hz为单位。
[0275]
在具有电喷雾电离(esi)离子源的混合四极杆飞行时间质谱仪(microtofq-ii,bruker daltonics,bremen)上以正离子模式记录hrms-esi质谱。对于低分辨率质谱,在thermo finnigan mat 95 xl光谱仪中记录lrms-esi质谱。
[0276]
第1部分:根据本发明的化合物的制备
[0277]
实施例1:4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0278]
n-(2-乙氧基乙基)-n,2-二甲基-丁-3-炔-2-胺的制备:在室温下向n-甲基-n-(2’羟基乙基)-3-氨基-3甲基-1-丁炔(easton,nelson r.;hennion,george f.u.s.(1967),us3337625 19670822.)(1.0g,7.08mmol)和碘乙烷(0.98ml,7.6mmol)的thf(12ml)溶液中
加入nah(0.459g,11.5mmol),并将混合物回流3h。然后在室温下用水小心地水解混合物,并用etoac(3x25ml)萃取。用盐水洗涤合并的有机层,用na2so4干燥并在真空下浓缩。通过硅胶色谱(石油醚/etoac=70/30)纯化粗品,得到纯的n-(2-乙氧基乙基)-n,2-二甲基-丁-3-炔-2-胺(0.479g,40%)。
[0279]1h nmr(300mhz,dmso)δ3.45-3.36(m,4h),3.11(s,1h),2.51(t,j=6.7hz,2h),2.20(s,3h),1.27(s,6h),1.09(t,j=7.0hz,3h)。
[0280]
esi-lrms 170.0[m h] 。
[0281]
4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯的制备:在-70℃下向n-(2-乙氧基乙基)-n,2-二甲基-丁-3-炔-2-胺(0.367g,2.17mmol)的thf(11ml)溶液中滴加2.28m n-buli的己烷溶液(1.14ml,2.60mmol)。在-70℃下5min后,将反应混合物升温至0℃,在该温度下保持10min然后在-70℃下冷却,之后用氧硫化碳(cos)鼓泡通过溶液30min。将黄色溶液升温至0℃,在该温度下再搅拌10min,然后滴加碘甲烷(0.162ml,2.60mmol)。将混合物搅拌2h,在0℃下用水小心地水解并用醚萃取。用盐水洗涤合并的有机层,用na2so4干燥并在真空下浓缩。通过硅胶色谱纯化粗品(石油醚/etoac=90/10)得到纯的4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯(0.369g,70%),为接近无色的油状物。
[0282]1h nmr(300mhz,dmso)δ3.42(t,j=6.3hz,2h),3.42(q,j=7.0hz,2h),2.56(t,j=6.3hz,2h),2.39(s,3h),2.25(s,3h),1.36(s,6h),1.10(t,j=7.0hz,3h)。
[0283]
esi-hrms c
12h22
no2s[m h] 计算值:244.1366,实测值:244.1362。
[0284]
实施例2:4-[2-烯丙基氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0285]
n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺的制备:在0℃下向n-甲基-n-(2’羟基乙基)-3-氨基-3甲基-1-丁炔(easton,nelson r.;hennion,george f.u.s.(1967),us3337625 19670822.)(1.0g,7.08mmol)的thf(12ml)溶液中加入nah(0.340g,8.50mmol)。在0℃下15min且在室温下15min后,在0℃下一批加入n-bu4ni(0.026g,0.071mmol),接着滴加烯丙基溴(0.735ml,8.50mmol)。将反应混合物升温至室温,搅拌过夜,然后用水小心地水解,并用醚萃取(3x25ml)。用盐水洗涤合并的有机层(25ml),用na2so4干燥并在真空下浓缩。通过硅胶色谱纯化(石油醚/醚=80/20至70/30)得到纯的n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺(0.941g,73%),为油状物。
[0286]1h nmr(300mhz,dmso)δ5.88(ddt,j=17.3,10.5,5.3hz,1h),5.24(ddd,j=17.3,3.8,1.7hz,1h),5.16-5.09(m,1h),3.93(dt,j=5.3,1.6hz,2h),3.43(t,j=6.4hz,2h),3.12(s,1h),2.55(t,j=6.4hz,2h),2.21(s,3h),1.27(s,6h)。
[0287]
esi-lrms 182.0[m h] 。
[0288]
4-[2-烯丙基氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯的制备:从n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:2.2mmol。通过硅胶色谱纯化(石油醚/etoac=90/10至80/20)。产率:65%。接近无色的油状物。
[0289]1h nmr(300mhz,dmso)δ5.88(ddt,j=17.3,10.5,5.3hz,1h),5.24(ddd,j=17.3,3.8,1.7hz,1h),5.17-5.10(m,1h),3.94(dt,j=5.3,1.5hz,2h),3.45(t,j=6.2hz,2h),
2.58(t,j=6.2hz,2h),2.39(s,3h),2.26(s,3h),1.36(s,6h)。
[0290]
esi-hrms c
13h22
no2s[m h] 计算值:256.1366,实测值:256.1364。
[0291]
实施例3:4-[2-苄基氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0292]
n-(2-苄基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺的制备:使用1.015当量nah和1.01当量溴苄,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=90/10)。产率:81%。无色油状物。
[0293]1h nmr(300mhz,dmso)δ7.39-7.24(m,5h),4.47(s,2h),3.49(t,j=6.3hz,2h),3.12(s,1h),2.58(t,j=6.3hz,2h),2.21(s,3h),1.27(s,6h)。esi-lrms 232.0[m h] 。
[0294]
4-[2-苄基氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯的制备:从n-(2-苄基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:2.2mmol。通过硅胶色谱纯化(石油醚/etoac=90/10)。产率:79%。无色油状物。
[0295]1h nmr(300mhz,dmso)δ7.37-7.26(m,5h),4.48(s,2h),3.52(t,j=6.1hz,2h),2.62(t,j=6.1hz,2h),2.38(s,3h),2.26(s,3h),1.36(s,6h)。
[0296]
esi-hrms c
17h24
no2s[m h] 计算值:306.1522,实测值:306.1514。
[0297]
可供选择的方案:在-70℃下向n-(2-苄基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺(0.650g,2.81mmol)的thf(8ml)溶液中滴加2.28m n-buli的己烷溶液(1.36ml,3.09mmol)。在-70℃下5min后,将反应混合物升温至0℃,在该温度下保持30min,并将co2鼓泡通过溶液30min。在5min内将混合物升温至室温然后在0℃下再冷却。滴加氯甲酸异丁酯(0.40ml,3.08mmol)并将混合物搅拌10min,之后一批加入甲醇钠(0.236g,3.37mmol)。将混合物升温至室温,在该温度下再搅拌15min,然后在0℃下用水小心地水解并用醚萃取。用盐水洗涤合并的有机层,用na2so4干燥并在真空下浓缩。通过硅胶色谱纯化粗品(石油醚/etoac=90/10)得到纯的4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯(0.307g,36%)。
[0298]
实施例4:4-甲基-4-[甲基-[2-(间甲苯基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯
[0299]
n-2-二甲基-n-[2-(间甲苯基甲氧基)乙基]丁-3-炔-2-胺的制备:用3-甲基溴苄,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=90/10)。规模:4.5mmol。产率:79%。无色油状物。
[0300]1h nmr(300mhz,dmso)δ7.26-7.19(m,1h),7.16-7.05(m,3h),4.43(s,2h),3.48(t,j=6.3hz,2h),3.12(s,1h),2.57(t,j=6.3hz,2h),2.30(s,3h),2.21(s,3h),1.27(s,6h)。
[0301]
esi-lrms 246.1[m h] 。
[0302]
4-甲基-4-[甲基-[2-(间甲苯基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯的制备:从n-2-二甲基-n-[2-(间甲苯基甲氧基)乙基]丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:1.3mmol。通过硅胶色谱纯化(石油醚/etoac=90/10)。产率:77%。
无色油状物。
[0303]1h nmr(300mhz,dmso)δ7.26-7.19(m,1h),7.16-7.05(m,3h),4.44(s,2h),3.50(t,j=6.1hz,2h),2.62(t,j=6.1hz,2h),2.38(s,3h),2.30(s,3h),2.26(s,3h),1.36(s,6h)。
[0304]
esi-hrms c
18h26
no2s[m h] 计算值:320.1679,实测值:320.1667。
[0305]
实施例5:4-[2-[(3,4-二甲基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0306]
n-[2-[(3,4-二甲基苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺的制备:用3,4-二甲基溴苄,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=60/40)。规模:3.8mmol。产率:60%。无色油状物。
[0307]1h nmr(300mhz,dmso)δ7.12-7.06(m,2h),7.04-6.99(m,1h),4.39(s,2h),3.45(t,j=6.3hz,2h),3.12(s,1h),2.56(t,j=6.3hz,2h),2.20(s,6h),2.19(s,3h),1.27(s,6h)。
[0308]
esi-lrms 182.0[m h] 。esi-lrms 260.0[m h] 。
[0309]
4-[2-[(3,4-二甲基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯的制备:从n-[2-[(3,4-二甲基苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:1.3mmol。通过硅胶色谱纯化(石油醚/etoac=90/10)。产率:77%。接近无色的油状物。
[0310]1h nmr(300mhz,dmso)δ7.12-7.06(m,2h),7.05-6.99(m,1h),4.40(s,2h),3.48(t,j=6.2hz,2h),2.60(t,j=6.2hz,2h),2.38(s,3h),2.25(s,3h),2.20(s,3h),2.19(s,3h),1.36(s,6h)。
[0311]
esi-hrms c
19h28
no2s[m h] 计算值:334.1835,实测值:334.1825。
[0312]
实施例6:4-[2-[(4-甲氧基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0313]
n-[2-[(4-甲氧基苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺的制备:用1-(溴甲基)-4-甲氧基苯,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(dcm/meoh=99/1至97.5/2.5)。规模:4.0mmol。产率:53%。无色油状物。
[0314]
1h nmr(300mhz,dmso)δ7.27-7.20(m,2h),6.93-6.86(m,2h),4.39(s,2h),3.74(s,3h),3.45(t,j=6.3hz,2h),3.12(s,1h),2.55(t,j=6.4hz,2h),2.20(s,3h),1.27(s,6h)。
[0315]
esi-lrms 261.9[m h] 。
[0316]
4-[2-[(4-甲氧基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯的制备:从n-[2-[(4-甲氧基苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:1.3mmol。通过硅胶色谱纯化(石油醚/etoac=80/20)。产率:74%。接近无色的油状物。
[0317]1h nmr(300mhz,dmso)δ7.27-7.21(m,2h),6.93-6.87(m,2h),4.40(s,2h),3.74(s,3h),3.48(t,j=6.2hz,2h),2.60(t,j=6.2hz,2h),2.38(s,3h),2.25(s,3h),1.36(s,6h)。
[0318]
esi-hrms c
18h26
no3s[m h] 计算值:336.1628,实测值:336.1613。
[0319]
实施例7:4-[2-[(3,4-二甲氧基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0320]
n-[2-[(3,4-二甲氧基苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺的制备:用4-(溴甲基)-1,2-二甲氧基苯,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物是。通过硅胶色谱纯化(dcm/meoh=99/1至97.5/2.5)。规模:4.0mmol。产率:67%。无色油状物。
[0321]1h nmr(300mhz,dmso)δ6.94-6.88(m,2h),6.87-6.80(m,1h),4.39(s,2h),3.74(s,3h),3.73(s,3h),3.46(t,j=6.3hz,2h),3.12(s,1h),2.56(t,j=6.3hz,2h),2.21(s,3h),1.27(s,6h)。
[0322]
esi-lrms 292.0[m h] 。
[0323]
4-[2-[(3,4-二甲氧基苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯的制备:从n-[2-[(3,4-二甲氧基苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:1.0mmol。通过硅胶色谱纯化(石油醚/etoac=90/10)。产率:67%。接近无色的油状物。
[0324]1h nmr(300mhz,dmso)δ6.94-6.87(m,1h),6.87-6.81(m,1h),4.40(s,2h),3.74(s,3h),3.73(s,3h),3.48(t,j=6.1hz,2h),2.61(t,j=6.2hz,2h),2.38(s,3h),2.26(s,3h),1.36(s,6h)。
[0325]
esi-hrms c
19h28
no4s[m h] 计算值:366.1734,实测值:336.1720。
[0326]
实施例8:4-[2-[(3-氯苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0327]
n-[2-[(3-氯苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺的制备:用3-氯溴苄,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=70/30)。规模:4.0mmol。产率:71%。无色油状物。
[0328]1h nmr(300mhz,dmso)δ7.43-7.25(m,4h),4.49(s,2h),3.50(t,j=6.2hz,2h),3.12(s,1h),2.58(t,j=6.2hz,2h),2.22(s,3h),1.28(s,6h)。
[0329]
esi-lrms 266.0[m h] 。
[0330]
4-[2-[(3-氯苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯的制备:从n-[2-[(3-氯苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物,除了在加入n-buli后将反应混合物在-70℃下保持30min,然后用cos鼓泡。规模:1.3mmol。通过硅胶色谱纯化(石油醚/etoac=75/25)。产率:63%。接近无色的油状物。
[0331]1h nmr(300mhz,dmso)δ7.44-7.24(m,4h),4.50(s,2h),3.52(t,j=6.0hz,2h),
2.63(t,j=6.0hz,2h),2.38(s,3h),2.27(s,3h),1.37(s,6h)。
[0332]
esi-hrms计算值c
17h23
clno2s[m h] :340.1133,实测值:340.1120。
[0333]
实施例9:4-[2-[(3-氟苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0334]
n-[2-[(3-氟苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺的制备:用3-氟溴苄,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=80/20)。规模:4.0mmol。产率:71%。无色油状物。
[0335]
4-[2-[(3-氟苯基)甲氧基]乙基-甲基-氨基]-4-甲基-戊-2-炔硫代酸s-甲酯的制备:从n-[2-[(3-氟苯基)甲氧基]乙基]-n,2-二甲基-丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物,除了在加入n-buli后将反应混合物在-70℃下保持30min,然后用cos鼓泡。规模:1.3mmol。通过硅胶色谱纯化(石油醚/etoac=70/30)。产率:87%。接近无色的油状物。
[0336]1h nmr(300mhz,dmso)δ7.44-7.34(m,1h),7.20-7.05(m,3h),4.51(s,2h),3.53(t,j=6.1hz,2h),2.63(t,j=6.1hz,2h),2.38(s,3h),2.27(s,3h),1.37(s,6h)。
[0337]
esi-hrms c
17h23
fno2s[m h] 计算值:324.1428,实测值:324.1415。
[0338]
实施例10:4-甲基-4-[甲基-[2-(2-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯
[0339]
n-2-二甲基-n-[2-(2-吡啶基甲氧基)乙基]丁-3-炔-2-胺的制备:从2-(溴甲基)吡啶氢溴酸盐开始,使用4当量nah,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(dcm/meoh=99/1至95/5)。规模:2.1mmol。产率:71%。黄色油状物。
[0340]1h nmr(300mhz,dmso)δ8.50(ddd,j=4.8,1.8,0.9hz,1h),7.80(td,j=7.7,1.8hz,1h),7.44(d,j=7.8hz,1h),7.32-7.24(m,1h),4.55(s,2h),3.56(t,j=6.2hz,2h),3.13(s,1h),2.61(t,j=6.2hz,2h),2.23(s,3h),1.28(s,6h)。
[0341]
esi-lrms 233.1[m h] 。
[0342]
4-甲基-4-[甲基-[2-(2-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯的制备:从n-2-二甲基-n-[2-(2-吡啶基甲氧基)乙基]丁-3-炔-2-胺开始,用1.5当量n-buli,1.5当量mei和dcm萃取,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:0.9mmol。通过硅胶色谱纯化(dcm/meoh=99/1至90/10)。产率:18%。黄色油状物。
[0343]1h nmr(300mhz,dmso)δ8.54-8.48(m,1h),7.80(td,j=7.7,1.8hz,1h),7.44(d,j=7.8hz,1h),7.28(dd,j=6.7,5.1hz,1h),4.56(s,2h),3.59(t,j=6.1hz,2h),2.65(t,j=6.1hz,2h),2.38(s,3h),2.28(s,3h),1.37(s,6h)。)。
[0344]
esi-hrms c
16h23
n2o2s[m h] 计算值:3071475,实测值:307.1471。
[0345]
实施例11:4-甲基-4-[甲基-[2-(3-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯
[0346]
n-2-二甲基-n-[2-(3-吡啶基甲氧基)乙基]丁-3-炔-2-胺的制备:从3-(溴甲基)
吡啶氢溴酸盐开始,使用4当量nah,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(dcm/meoh=99/1至95/5)。规模:2.1mmol。产率:67%。黄色油状物。
[0347]1h nmr(300mhz,dmso)δ8.56-8.52(m,1h),8.49(dd,j=4.8,1.7hz,1h),7.78-7.70(m,1h),7.38(ddd,j=7.8,4.8,0.8hz,1h),4.52(s,2h),3.52(t,j=6.2hz,2h),3.12(s,1h),2.58(t,j=6.2hz,2h),2.21(s,3h),1.27(s,6h)。
[0348]
esi-lrms 233.1[m h] 。
[0349]
4-甲基-4-[甲基-[2-(3-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯的制备:从n-2-二甲基-n-[2-(3-吡啶基甲氧基)乙基]丁-3-炔-2-胺开始,使用1.5当量n-buli,1.5当量mei和dcm萃取,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:0.4mmol。通过硅胶色谱纯化(dcm/meoh=99/1至95/5)。产率:15%。黄色油状物。
[0350]1h nmr(300mhz,dmso)δ8.54(d,j=1.5hz,1h),8.49(dd,j=4.8,1.7hz,1h),7.78-7.70(m,1h),7.38(ddd,j=7.8,4.8,0.8hz,1h),4.53(s,2h),3.54(t,j=6.1hz,2h),2.63(t,j=6.1hz,2h),2.38(s,3h),2.26(s,3h),1.36(s,6h))。
[0351]
esi-hrms c
16h23
n2o2s[m h] 计算值:3071475,实测值:307.1474。
[0352]
实施例12:4-甲基-4-[甲基-[2-(4-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯
[0353]
n-2-二甲基-n-[2-(4-吡啶基甲氧基)乙基]丁-3-炔-2-胺的制备:从4-(溴甲基)吡啶氢溴酸盐开始,使用4当量nah,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(dcm/meoh=99/1至95/5)。规模:2.1mmol。产率:95%。黄色油状物。
[0354]1h nmr(300mhz,dmso)δ8.56-8.49(m,2h),7.38-7.27(m,2h),4.54(s,2h),3.53(t,j=6.2hz,2h),3.13(s,1h),2.61(t,j=6.2hz,2h),2.23(s,3h),1.28(s,6h)。
[0355]
esi-lrms 233.1[m h] 。
[0356]
4-甲基-4-[甲基-[2-(4-吡啶基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯的制备:从n-2-二甲基-n-[2-(4-吡啶基甲氧基)乙基]丁-3-炔-2-胺开始,用1.5当量n-buli,1.5当量mei和dcm萃取,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:1.0mmol。通过硅胶色谱纯化(dcm/meoh=99/1至95/15)。产率:24%。黄色油状物。
[0357]1h nmr(300mhz,dmso)δ8.60-8.45(m,2h),7.37-7.26(m,2h),4.55(s,2h),3.56(t,j=6.0hz,2h),2.65(t,j=6.0hz,2h),2.38(s,3h),2.28(s,3h),1.37(s,6h)。
[0358]
esi-hrms c
16h23
n2o2s[m h] 计算值:3071475,实测值:307.1470。
[0359]
实施例13:4-(二甲基氨基)-4-甲基-戊-2-炔酸甲酯
[0360]
4-(二甲基氨基)-4-甲基-戊-2-炔酸盐酸盐的制备:在-70℃下向n,n,2-三甲基丁-3-炔-2-胺(0.928g,8.35mmol)的thf(42ml)溶液中滴加2.35m n-buli己烷溶液(3.73ml,8.76mmol)。在-70℃下5min后将反应混合物升温至0℃,在该温度下保持10min,然后在-70℃下冷却,之后用二氧化碳鼓泡45min。在2h内将混合物升温至0℃,然后在0℃下用水小心地水解并用醚洗涤(2x25ml)。用6n hcl酸化水层(ph1-2),然后真空浓缩。将得到的
烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(环己烷/etoac=80/20)。规模2.4mmol。产率:55%。黄色油状物。
[0381]1h nmr(300mhz,dmso)δ7.39-7.22(m,5h),4.44(s,2h),3.42(t,j=6.3hz,2h),3.08(s,1h),2.34(t,j=6.9hz,2h),2.13(s,3h),1.60-1.37(m,4h),1.26(s,6h)。
[0382]
esi-lrms:260.2[m h]
[0383]
4-((4-(苄基氧基)丁基)(甲基)氨基)-4-甲基戊-2-炔硫代酸s-甲酯的制备:从n-(4-苄基氧基丁基)-n,2-二甲基-丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(环己烷/etoac=90/10)。规模0.77mmol。产率:80%。黄色油状物。
[0384]
1h nmr(300mhz,cdcl3)δ7.38-7.19(m,5h),4.48(s,2h),3.47(t,j=6.1hz,2h),2.45(t,j=6.9hz,2h),2.35(s,3h),2.26(s,3h),1.73-1.48(m,4h),1.39(s,6h)。
[0385]
esi-hrms:c19h28no2s[m h] 计算值:334.1835,实测值334.1840。
[0386]
实施例18:4-((2-羟基乙基)(甲基)氨基)-4-甲基戊-2-炔硫代酸s-甲酯
[0387][0388]
a)见easton,nelson r.;hennion,george f.,u.s.(1966),us 3285913 b)3,4-dhp,ptsa,dcm(70%)c)nbuli,thf,-70℃,氧硫化碳然后mei 0℃(59%)d)ptsa,meoh,室温(90%)
[0389]
化合物3的制备:n,2-二甲基-n-(2-((四氢-2h-吡喃-2-基)氧基)乙基)丁-3-炔-2-胺:在室温下向2-(甲基(2-甲基丁-3-炔-2-基)氨基)乙醇2(3.00g,21.2mmol)和3,4-二氢-2h-吡喃(5.0当量)的无水dcm(135ml)溶液中加入对甲苯磺酸(0.1当量)。将反应混合物搅拌过夜,用饱和nahco3水溶液(30ml)洗涤然后用盐水(30ml)洗涤。用硫酸钠干燥有机层,过滤,并在真空下浓缩。首先用kugelrohr仪器(10-12托,烘箱155℃)通过短程蒸馏纯化残余物,然后通过快速硅胶色谱(石油醚/乙酸乙酯95/5至60/40)纯化,得到化合物3,为油状物(产率70%)。
[0390]1h nmr(300mhz,dmso)δ4.60-4.50(m,1h),3.82-3.70(m,1h),3.59-3.56(m,1h),3.49-3.35(m,2h),3.12(s,1h),2.55(t,j=6.5hz,2h),2.21(s,3h),1.77-1.35(m,6h),1.27(s,6h)。
[0391]
化合物4的制备:4-甲基-4-(甲基(2-((四氢-2h-吡喃-2-基)氧基)乙基)氨基)戊-2-炔硫代酸s-甲酯:向炔胺3(1.00g,4.44mmol)的无水thf(22ml)溶液中滴加n-丁基锂溶液(2.2m己烷溶液,1.5当量)。混合物在10分钟内达到0℃,然后再冷却至-70℃,然后用氧硫化碳鼓泡。30分钟后将亮黄色溶液小心地升温至0℃,在该温度下搅拌30分钟,并滴加碘甲烷(1.2当量)。将反应混合物在0℃下搅拌2小时,然后用水水解。用dcm萃取后处理(用盐水洗涤,用硫酸钠干燥并减压浓缩)得到粗品,将其通过硅胶色谱纯化(石油醚/乙酸乙酯90/10至60/40)得到化合物4,为油状物(产率59%)。
[0392]1h nmr(300mhz,dmso)δ4.58(t,j=3.2hz,1h),3.75(ddd,j=11.4,7.9,3.3hz,1h),3.70-3.60(m,1h),3.49-3.38(m,2h),2.59(t,j=6.3hz,2h),2.39(s,3h),2.27(s,3h),1.77-1.39(m,6h),1.36(s,6h)。
13
c nmr(75mhz,cdcl3)δ176.62(c=o),98.94(ch),96.46(c),80.97(c),66.55(ch2),62.37(ch2),55.19(c),52.43(ch2),37.92(ch3),30.75(ch2),28.01(2xch3),25.59(ch2),19.63(ch2),12.61(ch3)。
[0393]
化合物5的制备:4-((2-羟基乙基)(甲基)氨基)-4-甲基戊-2-炔硫代酸s-甲酯:在室温下向氨基硫醇酯4(1.00g,3.34mmol)的甲醇(15ml)溶液中加入对甲苯磺酸(1.1当量)。将反应混合物搅拌过夜,用饱和水溶液nahco3(30ml)洗涤,然后用盐水(30ml)洗涤。用硫酸钠干燥有机层,过滤,并在真空下浓缩。通过快速硅胶色谱(石油醚/乙酸乙酯80/20至20/80)纯化残余物得到化合物5,为油状物(产率90%)。
[0394]1h nmr(300mhz,dmso)δ4.42(t,j=5.6hz,1h),3.44(td,j=6.7,5.6hz,2h),2.46(,j=6.7hz,2h),2.39(s,3h),2.24(s,3h),1.36(s,6h)。
13
c nmr(75mhz,cdcl3)δ176.46(c=o),95.56(c),80.89(c),58.92(ch2),54.99(c),53.52(ch2),36.12(ch3),27.88(2xch3),12.53(ch3)。esi-hrms:c
10h18
no2s[m h]
计算值:216.1053,实测值:216.1043。
[0395]
实施例19:4-甲基-4-[甲基-[2-(2-萘基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯
[0396]
n,2-二甲基-n-[2-(2-萘基甲氧基)乙基]丁-3-炔-2-胺的制备:用1.015当量nah和1.01当量2-萘基溴,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=90/10)。产率:61%。橙色油状物。
[0397]
1h nmr(300mhz,dmso)δ7.91-7.88(m,4h),7.50-7.48(m,3h),4.65(s,2h),3.55(t,6.1hz,2h),3.13(s,1h),2.62(t,6.2hz,2h),2.23(s,3h),1.28(s,6h)。
[0398]
4-甲基-4-[甲基-[2-(2-萘基甲氧基)乙基]氨基]戊-2-炔硫代酸s-甲酯的制备:从n,2-二甲基-n-[2-(2-萘基甲氧基)乙基]丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。规模:0.65mmol。通过硅胶色谱纯化(石油醚/etoac=85/15)。产率:28%。黄色油状物。
[0399]
1h nmr(300mhz,dmso)δ7.95-7.82(m,4h),7.55-7.41(m,3h),4.66(s,2h),3.57(t,j=6.2hz,2h),2.66(t,j=6.1hz,2h),2.38(s,3h),2.28(s,3h),1.37(s,6h)。
[0400]
esi-hrms:c
21h26
no2s计算值356.1683,实测值356.1679[m h]
[0401]
实施例20:4-甲基-4-[甲基-[2-[(2,6,6-三甲基环己烯-1-基)甲氧基]乙基]氨基]戊-2-炔硫代酸s-甲酯
[0402]
n,2-二甲基-n-[2-[(2,6,6-三甲基环己烯-1-基)甲氧基]乙基]丁-3-炔-2-胺的制备:用1.015当量nah和1.01当量2-(溴甲基)-1,3,3-三甲基-1-环己烯(通过已知方案(wo2015048363),由β-环柠檬醛制备),通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=95/05)。产率:67%。浅黄色油状物。
[0403]
3.86(s,2h),3.41(t,j=6.5hz,2h),3.12(s,1h),2.53(t,j=6.4hz,2h),2.20(s,3h),1.90(t,j=5.9hz,2h),1.62(s,3h),1.57-1.49(m,2h),1.41-1.33(s,2h),1.27(s,
6h),0.97(s,6h)。
[0404]
s 4-甲基-4-[甲基-[2-[(2,6,6-三甲基环己烯-1-基)甲氧基]乙基]氨基]戊-2-炔硫代酸s-甲酯的制备:从n,2-二甲基-n-[2-[(2,6,6-三甲基环己烯-1-基)甲氧基]乙基]丁-3-炔-2-胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯(实施例19)所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=90/10)。产率:63%。黄色油状物。
[0405]
h nmr(300mhz,dmso)δ3.87(s,2h),3.44(t,j=6.3hz,2h),2.57(t,j=6.3hz,2h),2.38(s,3h),2.26(s,3h),1.91(t,j=6.2hz,2h),1.62(s,3h),1.57-1.49(m,2h),1.36(s,6h),1.39-1.34(m,2h),0.97(s,6h)。
[0406]
esi-hrms c20h34no2s[m h] 计算值:352.2305,实测值:352.2289。
[0407]
实施例21:乙酸2-[(1,1-二甲基-4-甲基硫烷基-4-氧代-丁-2-炔基)-甲基氨基]乙酯:
[0408]
在0℃下向4-[2-羟基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯(100mg,0.46mmol)和二异丙基乙胺(1.2当量)的二氯甲烷(2.3ml)溶液中滴加乙酰氯(1.2当量)。在0℃下10min后,将反应混合物升温至室温并搅拌至完全转化(tlc检查)。然后将混合物稀释在二氯甲烷(30ml)中,用盐水(40ml)洗涤,用硫酸钠干燥并减压蒸发溶剂。通过硅胶色谱(石油醚/乙酸乙酯75/25)纯化粗品得到产物21,为黄色油状物。产率58%。
[0409]
1h nmr(300mhz,dmso)δ4.05(t,j=6.1hz,2h),2.62(t,j=6.1hz,2h),2.39(s,3h),2.26(s,3h),2.01(s,3h),1.36(s,6h)。
[0410]
esi-hrms c12h19no3s[m h] 计算值:258.1086,实测值:258.1150。
[0411]
实施例22:3,4-二甲氧基苯甲酸2-[(1,1-二甲基-4-甲基硫烷基-4-氧代-丁-2-炔基)-甲基氨基]乙酯:
[0412]
从3,4-二甲氧基苯甲酰氯开始,通过使用与针对乙酸2-[(1,1-二甲基-4-甲基硫烷基-4-氧代-丁-2-炔基)-甲基氨基]乙酯[实施例21]所描述的相同的方法获得该化合物。规模:0.46mmol。通过硅胶色谱纯化(石油醚/etoac=60/40)。产率:《10%。黄色油状物。
[0413]
1h nmr(300mhz,dmso)δ7.59(dd,j=8.4,2.0hz,1h),7.45(d,j=2.0hz,1h),7.08(d,j=8.5hz,1h),4.28(t,j=7.1hz,2h),3.83(s,3h),3.80(s,3h),2.72(t,j=7.1hz,2h),2.38(s,3h),2.33(s,3h),1.39(s,6h)。
[0414]
实施例23:2,5,10,11,11-五甲基-6-氧代-7-氧杂-2,5,10-三氮杂十四碳-12-炔-14-硫代酸s-甲酯
[0415]
从n,n,n
’‑
三甲基乙二胺开始,通过使用与针对4-甲基-4-[甲基-[2-[甲基-[2-(甲基氨基)乙基]氨基甲酰基]氧基乙基]氨基]戊-2-炔硫代酸s-甲酯[化合物7/实施例29]所描述的相同的方法获得该化合物。通过硅胶色谱纯化dcm/meoh(85/15)。产率:34%。黄色油状物。
[0416]
1h nmr(300mhz,dmso)δ4.01(t,j=5.9hz,2h),3.32-3.25(m,2h),2.83(大s,3h),2.62(t,j=5.8hz,2h),2.6-2.5(m,被溶剂峰部分隐藏,2h),2.39(s,3h),2.27(s,3h),2.17(大s,6h),1.36(s,6h)。
[0417]
esi-hrms c16h30n3o3s[m h] 计算值:344.1992,实测值:344.1993。
[0418]
实施例24:(s)-4-(2-(甲氧基甲基)吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯
[0419]
(s)-2-(甲氧基甲基)-1-(2-甲基丁-3-炔-2-基)吡咯烷的制备:用1当量(s)-吡咯烷-2-基甲醇,1.015当量nah和1.01当量碘甲烷,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(二氯甲烷/甲醇=90/10)。规模3.0mmol。产率:61%。橙色油状物。
[0420]1h nmr(300mhz,dmso)δ3.23(s,3h),3.16-3.06(m,2h),3.05(s,1h),3.03-2.91(m,1h),2.91-2.80(m,1h),2.67-2.54(m,1h),1.77-1.49(m,4h),1.30(s,3h),1.24(s,3h)。
[0421]
esi-lrms:182.2[m h] 。
[0422]
(s)-4-(2-(甲氧基甲基)吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯的制备:从(s)-2-(甲氧基甲基)-1-(2-甲基丁-3-炔-2-基)吡咯烷开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=80/20)。规模0.8mmol。产率:57%。黄色油状物。
[0423]1h nmr(300mhz,dmso)δ3.24(s,3h),3.19-2.89(m,4h),2.67-2.53(m,1h),2.38(s,3h),1.83-1.50(m,4h),1.39(s,3h),1.33(s,3h)。
[0424]
esi-hrms计算值c
13h22
no2s[m h] :256.1366,实测值:256.1363。
[0425]
实施例25:4-[(3r)-3-甲氧基吡咯烷-1-基]-4-甲基-戊-2-炔硫代酸s-甲酯
[0426]
(r)-3-甲氧基-1-(2-甲基丁-3-炔-2-基)吡咯烷的制备:从(r)-1-(2-甲基丁-3-炔-2-基)吡咯烷-3-醇开始,使用1.015当量nah和1.01当量碘甲烷,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(二氯甲烷/甲醇=90/10)。规模3.3mmol。产率:30%。黄色油状物。
[0427]1h nmr(300mhz,dmso)δ3.85(ddd,j=10.8,7.2,3.6hz,1h),3.17(s,3h),3.13(s,1h)2.82(dd,j=9.8,6.5hz,1h),2.66-2.60(m,1h),2.56-2.50(m,2h),2.01-1.87(m,1h),1.73-1.55(m,1h),1.28(s,6h)。
[0428]
esi-lrms:168.0[m h] 。
[0429]
(r)-4-(3-甲氧基吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯的制备:
[0430]
从(r)-3-甲氧基-1-(2-甲基丁-3-炔-2-基)吡咯烷开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=60/40)。规模0.8mmol。产率:55%。黄色油状物。
[0431]1h nmr(300mhz,dmso)δ3.87(ddd,j=10.5,6.9,3.4hz,1h),3.18(s,3h),2.90-2.76(m,1h),2.77-2.65(m,1h),2.65-2.54(m,2h),2.38(s,3h),2.07-1.86(m,1h),1.76-1.60(m,1h),1.36(s,6h)。
[0432]
实施例26:4-(((1r,2r)-2-(苄基氧基)环戊基)(甲基)氨基)-4-甲基戊-2-炔硫代酸s-甲酯
[0433]
(1r,2r)-2-苄基氧基-n-(1,1-二甲基丙-2-炔基)环戊胺的制备:
[0434]
在室温下向市售的(1r,2r)-2-(苄基氧基)环戊胺(0.93g,4.86mmol),3-氯-3-甲基丁-1-炔(1.3当量)和三乙胺(1.3当量)的thf(20ml)溶液中加入cui(8mol%)。将混合物搅拌过夜。减压蒸发溶剂并将粗品稀释于饱和nahco3水溶液中,用乙酸乙酯萃取。用2%nh4oh水溶液洗涤合并的有机层,然后用盐水洗涤,用na2so4干燥并减压蒸发溶剂。得到(1r,2r)-2-苄基氧基-n-(1,1-二甲基丙-2-炔基)环戊胺,为棕色油状物。产率:99%。
[0435]1h nmr(300mhz,dmso)δ7.37-7.16(m,5h),4.56-4.36(m,2h),3.72-3.58(m,1h),3.28-3.19(m,1h),3.08(s,1h),2.00-1.88(m,1h),1.86-1.67(m,2h),1.65-1.49(m,3h),1.40-1.28(m,1h),1.25(s,3h),1.25(s,3h)。
[0436]
(1r,2r)-2-(苄基氧基)-n-甲基-n-(2-甲基丁-3-炔-2-基)环戊胺的制备:向(1r,2r)-2-(苄基氧基)-n-(2-甲基丁-3-炔-2-基)环戊胺(0.25g,0.97mmol)加入5当量甲酸和1.5当量甲醛(37%水溶液)。将混合物回流过夜,然后加入2n hcl直至达到ph 1,并用醚洗涤。用1n naoh碱化水层,并用dcm萃取。用盐水洗有机层,用na2so4干燥并减压蒸发溶剂。然后通过硅胶色谱(石油醚/etoac=80/20)纯化粗品,得到黄色油状物。产率:68%。
[0437]1h nmr(300mhz,dmso)δ7.43-7.17(m,5h),4.52(m,2h),3.94-3.78(m,1h),3.62-3.49(m,1h),3.11(s,1h),2.20(s,3h),1.78-1.45(m,6h),1.37(s,3h),1.34(s,3h)。
[0438]
esi-lrms:272.1[m h] 。
[0439]
4-(((1r,2r)-2-(苄基氧基)环戊基)(甲基)氨基)-4-甲基戊-2-炔硫代酸s-甲酯的制备:从(1r,2r)-2-(苄基氧基)-n-甲基-n-(2-甲基丁-3-炔-2-基)环戊胺开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=80/20然后dcm=100)。规模0.64mmol。产率:47%。黄色油状物。
[0440]1h nmr(300mhz,dmso)δ7.35-7.20(m,5h),4.51(s,2h),3.92-3.80(m,1h),3.59-3.45(m,1h),2.36(s,3h),2.24(s,3h),1.78-1.49(m,6h),1.45(s,3h),1.42(s,3h)。
[0441]
esi-hrms c
20h28
no2s[m h] 计算值:346.1835,实测值:346.1824。
[0442]
实施例27:(s)-4-(2-((苄基氧基)甲基)吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯:
[0443]
(s)-2-((苄基氧基)甲基)-1-(2-甲基丁-3-炔-2-基)吡咯烷的制备:从(s)-(1-(2-甲基丁-3-炔-2-基)吡咯烷-2-基)甲醇,1.015当量nah和1.01当量溴苄开始,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(二氯甲烷/甲醇=95/5)。规模3.0mmol。产率:60%。橙色油状物。
[0444]1h nmr(300mhz,dmso)δ7.41-7.19(m,5h),4.46(s,2h),3.24(dd,j=8.3,2.7hz,1h),3.14(dd,j=7.6,3.2hz,1h),3.11-3.02(m,2h),2.86(dd,j=8.4,6.0hz,1h),2.65-2.54(m,1h),1.81-1.51(m,4h),1.25(s,3h),1.23(s,3h)。
[0445]
esi-lrms:258.1[m h] 。
[0446]
(s)-4-(2-((苄基氧基)甲基)吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯的制备:从(s)-2-((苄基氧基)甲基)-1-(2-甲基丁-3-炔-2-基)吡咯烷开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(石油醚/etoac=80/20)。规模1.2mmol。产率:50%。橙色油状物。
[0447]1h nmr(300mhz,dmso)δ7.46-7.18(m,5h),4.47(s,2h),3.29-3.23(m,1h),3.17-3.03(m,2h),2.94(dd,j=8.5,5.7hz,1h),2.62-2.53(m,1h),2.38(s,3h),1.85-1.58(m,4h),1.34(s,3h),1.32(s,3h)。
[0448]
实施例28:(r)-4-(3-(苄基氧基)吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯:
[0449]
(r)-3-(苄基氧基)-1-(2-甲基丁-3-炔-2-基)吡咯烷的制备:从(r)-1-(2-甲基丁-3-炔-2-基)吡咯烷-3-醇开始,用1.01当量nah和1.01当量溴苄,通过使用与针对n-(2-烯丙基氧基乙基)-n,2-二甲基-丁-3-炔-2-胺[实施例2]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(二氯甲烷/甲醇=98/2)。规模3.3mmol。产率:69%。橙色油状物。
[0450]1h nmr(300mhz,dmso)δ7.40-7.17(m,5h),4.42(s,2h),4.07(tt,j=7.3,3.7hz,1h)3.13(s,1h),2.86(dd,j=9.6,6.7hz,1h),2.75-2.65(m,2h),2.60-2.53(m,1h),2.06-1.95(m,1h),1.75-1.65(m,1h),1.29(s,6h)。
[0451]
esi-lrms 244.1[m h] 。
[0452]
(r)-4-(3-(苄基氧基)吡咯烷-1-基)-4-甲基戊-2-炔硫代酸s-甲酯的制备:从(r)-3-(苄基氧基)-1-(2-甲基丁-3-炔-2-基)吡咯烷开始,通过使用与针对4-[2-乙氧基乙基(甲基)氨基]-4-甲基-戊-2-炔硫代酸s-甲酯[实施例1]所描述的相同的方法获得该化合物。通过硅胶色谱纯化(二氯甲烷/甲醇=98/2)。规模1.2mmol。产率:69%。橙色油状物。
[0453]1h nmr(300mhz,dmso)δ7.32(m,5h),4.44(s,2h),4.09(dq,j=10.2,3.5hz,1h),2.97-2.83(m,1h),2.80-2.65(m,2h),2.65-2.55(m,1h),2.37(s,3h),2.11-1.90(m,1h),1.83-1.67(m,1h),1.37(s,6h)。
[0454]
esi-hrms c
18h24
no2s[m h] 计算值:318.1522,实测值:318.1518。
[0455]
实施例29:adc化合物(无抗体)
[0456]
接头(mc-val-cit-pab-pnp)的制备:用于缀合物制备的所选接头是在已用于利妥昔单抗和其他的已知平台上设计的,包括用于连接至抗体的马来酰亚胺,组织蛋白酶可切割基团和用于1,6-消除的对氨基苄基系统:mc-val-cit-pab-pnp,cas 159857-81-5。
[0457]
它是按照从fmoc-val-osu[cas 3392-12-9]开始的标准方案制备的,或者可以购自商业供应商(例如creative biolabs,alb technology,carbosynth等)。
[0458]
接头的通式如下所示:
[0459][0460]
与mc-val-cit-pab-pnp偶联的根据本发明的化合物(实施例18)的制备
[0461]
化合物6:
[0462]
在0℃下向对硝基氯甲酸酯(140mg,0.70mmol,1.5当量)的dcm(2.5ml)溶液中滴加化合物5(100mg,0.46mmol)和tea(1.5当量)的dcm(1.5ml)溶液。在0℃下10分钟后,将反应混合物升温至室温并搅拌至完全转化(tlc检查,1h)。然后将混合物稀释于dcm(30ml)中,用盐水(40ml)洗涤,用na2so4干燥并减压蒸发溶剂。通过硅胶色谱(石油醚/乙酸乙酯100/至70/30)纯化粗品得到化合物6,为无定形固体(产率82%)。1h nmr(300mhz,dmso):δ(ppm)8.35-8.30(m,2h),7.59-7.53(m,2h),4.30(t,j=5.7hz,2h),2.75(t,j=5.7hz,2h),2.39(s,3h),2.30(s,3h),1.39(s,6h)。
[0463]
化合物7:
[0464]
在室温下向化合物6(200mg,0.53mmol)的dcm(3ml)溶液中加入tea(1.2当量)。然后在0℃下加入n-甲基-n-[2-(甲基氨基)乙基]氨基甲酸叔丁酯(1.2当量)的dcm(2.3ml)溶液。将得到的亮黄色反应混合物升温至室温并搅拌过夜。将混合物稀释于30ml dcm中,用40ml盐水洗涤,用na2so4干燥并减压蒸发溶剂。通过硅胶色谱(石油醚/乙酸乙酯75/25至20/80)纯化粗品,得到化合物7,为粘稠油状物(产率70%)。
[0465]1h nmr(300mhz,dmso):δ(ppm)3.99(t,j=5.7hz,2h),3.30(s,3h),2.85-2.70(m,7h),2.66-2.58(m,2h),2.38(s,3h),2.26(s,3h),1.37(s,9h),1.36(s,6h)。esi-lrms:c
20h36
n3o5s[m h]
计算值:430.2;实测值:430.2。
[0466]
化合物8:
[0467]
在0℃下向化合物7(44.5mg,0.104mmol)的4.5ml dcm(4.5ml)溶液中加入tfa(0.5ml)。然后将混合物升温至室温并搅拌过夜。真空浓缩后(浴t℃《45℃),在et2o中研磨和超声处理油状残余物。用et2o洗涤得到的粘性固体(双三氟乙酸盐)并干燥。产率:67%。
[0468]1h nmr(300mhz,d2o)δ4.57-4.44(m,2h),3.73-3.69(m,2h),3.62(t,j=5.7hz,2h),3.30-3.19(t,j=5.8hz,2h),3.05-2.99(m,3h),2.99-2.90(m,3h),2.73(s,3h),2.45(s,3h),1.79(s,6h)。esi-lrms:计算值c
15h28
n3o3s[m h]
:330.2;实测值:330.1。
[0469]
[0470]
化合物9:
[0471]
在0℃下向mc-val-cit-pabc-pnp(52mg,0.07mmol)和化合物8(1.06当量)的dmf(1.4ml)溶液中依次加入:一批加入hobt(1当量),然后滴加dipea(3当量)。在0℃下5分钟后,将混合物升温至室温并搅拌过夜。然后真空浓缩混合物(蒸发掉3/4,浴t℃《45℃)并用et2o使其絮凝。在et2o中洗涤/研磨(x3)后得到白色粗固体。通过硅胶色谱(meoh/dcm 95/5至90/10%)最终纯化得到终产物(化合物9),为白色固体。产率:75%。
[0472]1h nmr(300mhz,dmso)δ9.99(s,1h),8.08(d,j=7.4hz,1h),7.80(d,j=8.6hz,1h),7.59(d,j=8.5hz,2h),7.35-7.22(m,2h),7.01(s,2h),5.97(t,j=5.8hz,1h),5.41(s,2h),4.98(s,2h),4.44-4.32(m,1h),4.25-4.14(m,1h),3.99(s,2h),3.45-3.33(m,4h),3.09-2.89(m,2h),2.89-2.70(m,6h),2.65-2.56(m,2h),2.38(s,3h),2.30-2.11(m,5h),2.10-1.90(m,1h),1.77-1.09(m,18h),0.84(dd,j=9.5,6.8hz,6h)。
[0473]
lc:zorbax,acn/h2o 0.1%tfa,254nm,96%。
[0474]
esi-hrms:c
44h66
n9o
11
s[m h]
计算值928.4597实测值928.4586。
[0475]
第2部分:根据本发明的化合物的用途
[0476]
实施例1:根据本发明的化合物的活性
[0477]
材料与方法
[0478]
细胞系。白血病细胞系hl-60(来自一名患有aml-m2的36岁女性)用于确定药物功效。细胞获自欧洲细胞培养物保藏中心(european collection of cell cultures,ecacc)。根据供应商的建议,所有细胞都在适宜的培养基中培养。
[0479]
细胞生存力测定,96孔形式。细胞生存力测定,96孔形式。以确保在实验结束时在对照(未处理的细胞)中约80%汇合所需的浓度将细胞接种到96孔细胞培养板中(0.5 x 10
4-5 x 104个细胞/孔)。
[0480]
使用范围从0.5到100μm(0.5,1,2,5,10,15,20,25,30,40,50,100μm)的每种化合物的不同浓度确定对根据本发明的化合物的敏感性。在37℃下在含5%co2的潮湿气氛中孵育48小时后,根据制造商的说明,使用刃天青(resazurin)分析化合物的生长抑制作用。
[0481]
为了确保良好的数据质量并将移液错误的影响降至最低,每个化合物浓度均基于8个独立孔的平均荧光强度进行评估。通过每个特定细胞系的半数最大抑制浓度(ic
50
)量化化合物反应,并通过对数剂量/反应曲线的非线性回归分析确定。
[0482]
统计分析。值表示为平均值
±
sd或频率和比例。使用四参数回归线确定细胞生存力曲线。在适当的情况下,通过非配对t检验,卡方检验(chi-square),fisher精确检验或anova确定组间差异。认为p《0.05具有统计学意义。使用graphpad prism 5.0版(graphpad software,san diego california usa)进行分析。
[0483]
结果
[0484]
通过测试根据本发明的化合物在hl60细胞中的细胞毒活性获得的ic
50
见下表3。
[0485]
表3
[0486]
[0487][0488]
实施例2:化合物3和5在人重组aldh1a1,aldh1a2,aldh1a3,aldh2和aldh3a1酶促分析中的活性
[0489]
材料与方法
[0490]
以1mg/ml制备人重组aldh1a1,aldh1a2,aldh1a3,aldh2(creative biomart,ny,usa)。使用饱和浓度的底物进行酶促反应。为了测试酶的酶活性,在不同测试化合物存在或
不存在的情况下,将10μl酶添加到含有50mm hepes ph 7.2,30mm mgcl2加上20mm nad 辅因子和2mm己醛的反应缓冲液(sigma-aldrich,st.louis,missouri,usa)中。用烟酰胺腺嘌呤二核苷酸还原形式(nadh,500μm,sigma-aldrich)在反应缓冲液(50mm hepes ph 7.2,30mm mgcl2)中制备内标。
[0491]
对于aldh3a1(1mg/ml),将10μl酶添加到含有50mm tris,5mm dtt,ph 8加上40mm烟酰胺腺嘌呤二核苷酸磷酸(氧化形式,nadp )和4-硝基苯甲醛(4-nba)的反应缓冲液(sigma-aldrich)中。用烟酰胺腺嘌呤二核苷酸磷酸还原形式(nadph,5μm,sigma-aldrich)在反应缓冲液(50mm tris,5mm dtt,ph 8)中制备内标。
[0492]
在37℃下在1ml石英比色皿中进行0-2-5-10-15-20-30-45-60分钟的时间依赖性抑制分析。监测nadh的形成,使用cary eclipse varian荧光计在激发波长=340nm/发射波长=460nm(荧光)下读取样品至少600秒。
[0493]
阴性对照由相同的反应组成,但不添加酶(酶空白)。为了确定酶空白的斜率并计算产物浓度(荧光单位),使用了以下公式:
[0494][0495]
其中cst是标准nadh浓度,fst是标准荧光,df/dt是时间相关荧光的斜率(s.等,2012)。
[0496]
在抑制剂不存在的情况下,酶的比活性(μmol/min
·
mg-u/mg)如下计算:
[0497][0498][0499]
在所述反应中,将根据本发明的化合物3和5的活性与dimate(4-(二甲基氨基)-4-甲基戊-2-炔硫代酸s-甲酯,描述于ep1296946中)的活性进行比较。
[0500]
结果
[0501]
获得的ic
50
见下表4。
[0502]
表4
[0503][0504]
化合物3和5显示出比dimate更高的对aldh 1类酶的抑制作用。
[0505]
实施例3:化合物3和5在与人重组aldh1a1,aldh1a2,aldh1a3,aldh2或aldh3a1反应中的动力学参数
[0506]
材料与方法
[0507]
动力学数据表示为三次独立测定的平均值
±
标准误差。kinact/ki由kobs对抑制剂浓度[i]图确定。kobs是根据酶与不同浓度的化合物3和5或dimate(即300μm,200μm,100μm,50μm,20μm和10μm的抑制剂)的产物浓度对孵育时间的图确定的。使用非线性回归程序grafit 5.0(erithacus software)由负指数拟合得到kobs。
[0508]
结果
[0509]
获得的动力学参数见下表5。
[0510]
表5
[0511][0512]
化合物3和5对aldh 1类重组酶,特别是aldh1a2和aldh1a3的抑制效力比dimate高4到20倍。
[0513]
实施例4:化合物3和5的全酶和生化表征后的抑制类型
[0514]
材料与方法
[0515]
为了确定测试化合物对每种同工酶的抑制机制,如上所述计算相应的kobs,将酶与300μm,200μm,100μm,50μm,20μm和10μm的测试化合物的产物浓度对孵育时间作图。使用非线性回归程序grafit 5.0(erithacus software)由负指数拟合得到每个测试抑制剂浓度的k
obs
。最后,由k
obs
对相应抑制剂浓度的图确定k
inact
/ki的动力学参数。数据线性拟合的斜率表示以μm-1
·
min-1
为单位的速率常数。基于得到的不同的图,将不可逆抑制表征为特
异性或非特异性亲和标记。
[0516]
在特异性亲和力标记的情况下,该图显示了饱和kobs与抑制剂浓度的关系图(类似于可逆抑制图),在高浓度抑制剂下达到了平台。在非特异性亲和标记中,k
obs
对抑制剂浓度的依赖性表现为非饱和。
[0517]
在所述实验中,将化合物3和5与dimate进行了比较。
[0518]
结果
[0519]
获得的结果见下表6。
[0520]
表6
[0521][0522]
尽管所有表征的化合物都显示出不可逆的抑制作用,但观察到的不同同工酶的失活类型在特异性和非特异性亲和标记之间有所不同。值得注意的是,化合物3和5以更高的特异性与aldh1a3相互作用,而对于dimate,抑制是通过如针对非特异性亲和标记,即不可逆抑制剂所描述的单步失活机制发生的。
[0523]
第3部分:根据本发明的抗体药物缀合物(adc)的制备
[0524]
adc是三组分系统,包含通过可生物降解的接头与抗体连接的细胞毒剂。抗体与癌细胞表面的特定标志物(抗原或受体)结合。然后整个抗体-药物缀合物被内化到癌细胞内,其中接头被降解并释放活性药物。
[0525]
在本发明的上下文中,细胞毒剂,即根据本发明的化合物,通过连接基团(马来酰亚胺,琥珀酰亚胺酯,酶的特定肽序列底物等)与抗体偶联,连接至可切割的接头(蛋白酶位点,肼,二硫化物)或不可切割,并且具有或不具有自牺牲间隔基团。
[0526]
根据本发明的化合物的合成
[0527]
参考第1部分实施例29。
[0528]
与抗体利妥昔单抗偶联
[0529]
在37℃下将抗体利妥昔单抗与dtt混合30分钟,然后使用amicon ultra-15,mwco 30kda,merck-millipore对含有1mmol/l edta的pbs进行渗滤。硫醇浓度通过ellman试剂5,5
’‑
二硫代-双(2-硝基苯甲酸)[dtnb]定量。在4℃下将溶解在dmf中的前述段落中获得的50倍摩尔过量的最终化合物9添加到还原的抗体中1小时。使用amicon ultra-15,mwco 30kda,merck-millipore在pbsx1中对抗体-药物缀合物进行渗滤。为了确定药物抗体比(dar),通过ellman试剂定量偶联后修饰抗体的硫醇浓度。
[0530]
根据本发明的化合物的释放机制,随后是组织蛋白酶切割基团,如下图所示:
[0531][0532]
释放的片段:
[0533][0534]
第4部分:根据本发明的adc的用途
[0535]
药物/抗体比(dar)的确定。
[0536]
在通过二硫苏糖醇(dtt)对利妥昔单抗进行轻度硫醇化并通过马来酰亚氨基团的偶联淬灭这些游离硫醇之后,通过使用ellman试剂的硫醇定量之间的差异控制第3部分中提到的抗体-药物缀合物的dar。dtt硫醇化后,利妥昔单抗分子产生了10个新的游离硫醇基团。在最终产物9偶联后,淬灭所有这些新的游离硫醇基团,使得每个利妥昔单抗分子偶联10个化合物9。
[0537]
利妥昔单抗-化合物9的细胞毒性。
[0538]
将50000个活的raji细胞一式三份铺板。然后,加入利妥昔单抗-化合物9的1:2连续稀释液或对照利妥昔单抗以产生最终浓度(起始浓度50μg/ml)。将细胞孵育48小时,此时向每个孔中加入20μl阿尔玛蓝(alamar blue)(thermo fisher scientific)。将板再孵育四小时,并使用540nm的激发波长和620nm的发射波长在读板器上读取荧光强度。
[0539]
结果表明,使用500μg/ml的利妥昔单抗-化合物9处理48小时的raji细胞生存力显著低于使用利妥昔单抗本身的raji细胞生存力(图1;p值《0.01;**)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。