1.本发明涉及合成化学技术领域,尤其涉及一种连续流制备羟丙基吡喃三醇的合成方法以及合成系统。
背景技术:
2.羟丙基吡喃三醇,又名玻色因,英文名:pro-xylane,cas号:439685-79-7,是一种具有生物活性的化妆品原料。羟丙基吡喃三醇可以通过激活黏多糖(gags)的合成,促进生成透明质酸和胶原蛋白;另外,还可通过改善真皮与表皮间的粘合度,诱导真皮和表皮结构成分的合成,促进受损组织的再生,帮助维持真皮的弹性,预防皮肤老化,因此,羟丙基吡喃三醇具有抗衰老、抗脱水等生物活性。研究表明,羟丙基吡喃三醇易于生物降解,不会在生物体内积累,因此,没有毒性,被认为是抗衰新秀。
[0003][0004]
对于羟丙基吡喃三醇的合成方法,目前虽有一些文献报道,但往往需要柱层析,后处理工艺复杂,成本较高。
[0005]
cavezzaa,boulle c,gu
é
guiniata等人在期刊[bioorganic&medicinal chemistry letters,2009,19(3):845-849]最早报道了以木糖为原料,在碱性条件下,与乙酰丙酮缩合反应12h,经强酸性阳离子交换树脂酸化,再用硼氢化物对羰基还原反应12h,合成羟丙基吡喃三醇。反应采用碳酸氢钠做碱,反应时间长,收率低。
[0006]
philippe等人在期刊[carbohydr chem,2014,40:1-10]中报道,以木糖为原料,在碳酸氢钠作用下,与乙酰丙酮进行缩合反应,转换为c-糖苷,再用重金属催化剂ru/c对羰基还原,合成羟丙基吡喃三醇。需要用到重金属ru,无法保证重金属残留,对产品质量有影响。
[0007]
黄冬婷等在期刊[广东化工,2018,45(10):73-74]中报道,以木糖为原料,采用强碱性阴离子树脂代替了普通的无机碱,第二步再用硼氢化钠对羰基进行还原,合成羟丙基吡喃三醇。反应液需要浓缩水,并柱层析产品,收率仅50%,不适合工业化。
[0008]
王长斌等在期刊[食品与药品,2020,22(6),498-499]中报道,以木糖为原料,采用氢氧化钾做碱,再以三乙酰氧基硼氢化钠做还原剂合成羟丙基吡喃三醇。三乙酰氧基硼氢化钠的还原能力弱,需要过量还原剂,反应时间长,成本高,反应制备的产品有醋酸味道,影响产品质量。
[0009]
专利cn201910785216.6报道,以木糖和乙酰乙酸乙酯为原料,在稀土金属配合物促进下,一锅法合成羟丙基吡喃三醇的方法。反应用稀土金属催化,重金属残留会影响产品品质,而且引入有机催化剂,未增加除杂工艺,对产品质量有影响。
[0010][0011]
除用化学合成羟丙基吡喃三醇外,也有生物合成法制备羟丙基吡喃三醇的报道,例如:cn202010629023.4报道了采用生物酶法合成羟丙基吡喃三醇。以木糖和异丙醇作为底物,以筛选得到的异丙醇脱氢酶、羟丙基吡喃三醇合成酶、羰基还原酶作为催化剂一锅法合成羟丙基吡喃三醇。在后处理过程中分别用超滤的方法除酶,用纳滤浓缩,用甲苯萃取等多步操作工序,工艺复杂。
[0012]
另外,专利202110383018.4公开了一种一锅法合成玻色因的方法,虽然该方法的步骤相对简单,但是获得的产品为淡黄色/黄色油状物,且有浓烈的醋酸刺激味道,不利于化妆品生产使用。
技术实现要素:
[0013]
为了克服现有技术的不足,本发明的目的之一在于提供一种连续流制备羟丙基吡喃三醇的合成方法,所得羟丙基吡喃三醇纯度好,焦糖等杂质少,刺激性气味少,异构体比例少,单一构型占比高。
[0014]
本发明的目的之二在于提供一种连续流制备羟丙基吡喃三醇的合成系统。
[0015]
本发明的目的之一采用如下技术方案实现:
[0016]
一种连续流制备羟丙基吡喃三醇的合成方法,使用连续流工艺,反应过程中所得中间体无需分离,包括以下步骤:木糖和乙酰化试剂在碱性条件下,在连续流反应器中进行取代反应和还原反应,接着经过淬灭后再进行萃取、分液,脱溶后即得。所得羟丙基吡喃三醇的结构式如下式的ⅰ所示。
[0017][0018]
更加具体地,反应流程如下:将式ii化合物d-木糖溶解于水或有机溶剂中,在碱液存在下,与乙酰化试剂(乙酰丙酮或乙酰乙酸乙酯)在微反应器中混合反应,所得中间体无需分离,接着直接于第二反应器中与还原试剂还原,并经过淬灭后连续分液,脱溶后可得高浓度产品式i化合物,反应路线如下:
[0019][0020]
本发明所提供的连续流制备羟丙基吡喃三醇的合成方法,能够克服现有制备技术存在的安全环保和空间收率问题,减少杂质生成,可控制单一构型比例,有利于提高产品活
性。同时,该方法原料廉价易得、反应条件温和、操作简便、合成效率高、杂质含量低、对环境友好,适合规模生产。
[0021]
进一步地,一种连续流制备羟丙基吡喃三醇的合成方法,使用连续流工艺,包括以下步骤:
[0022]
第一混合步骤:木糖溶液、酰化试剂溶液、以及碱性溶液分别通过木糖溶液进料系统、酰化试剂溶液进料系统以及碱性溶液进料系统泵入第一混合器中,混合,得到第一混合物;
[0023]
取代反应步骤:所述第一混合物进入第一反应器中进行取代反应,反应一定时间后,得到第二混合物;
[0024]
第二混合步骤:所述第二混合物进入第二混合器,并向所述第二混合器泵入还原剂,反应一定时间后,得到第三混合物;
[0025]
还原反应步骤:所述第三混合物进入第二反应器中进行还原反应,得到第四混合物;
[0026]
淬灭反应步骤:所述第四混合物进入第三混合器中进行酸淬灭反应,得到第五混合物;
[0027]
分离步骤:所述第五混合物进入油水连续分离器中进行萃取、分液,然后脱溶,即得。
[0028]
进一步地,在所述第一混合步骤中,所述木糖溶液的溶质为木糖,溶剂为水、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚、丙酮、乙腈、甲醇、乙醇、异丙醇、正丁醇、乙二醇、丙三醇、1,3-丁二醇中的一种或任意组合,优选为水、甲醇、乙醇或正丁醇。
[0029]
进一步地,所述酰化试剂溶液中,酰化试剂为乙酰丙酮和/或乙酰乙酸乙酯,溶解酰化试剂的溶剂为四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚、丙酮、乙腈、甲醇、乙醇、异丙醇、正丁醇、甲苯、二甲苯、氯苯、二氯甲烷中的一种或任意组,优选为四氢呋喃、甲醇、乙醇或正丁醇。
[0030]
进一步地,所述碱性溶液中所用的碱为氢氧化钠、氢氧化锂、氢氧化钾、碳酸钠、碳酸钾、碳酸铯、氢氧化镁、氢氧化钙、碳酸氢钠、碳酸氢钾、磷酸钠、磷酸钾、磷酸氢二钠、磷酸氢二钾中的一种或任意组合,优选为氢氧化钠、氢氧化钾或碳酸钠。
[0031]
进一步地,在所述第二混合步骤中,所述还原剂为硼氢化钠、硼氢化钾、三乙酸硼氢化钠、氰基硼氢化钠、氢化铝锂中的一种或任意组合;溶解或悬浮所述还原剂的溶剂为水、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚、丙酮、乙腈、甲醇、乙醇、异丙醇、正丙醇、正丁醇、乙二醇、1,3-丁二醇中的一种或任意组合,优选为水、甲醇、乙醇或正丁醇。
[0032]
进一步地,在所述淬灭反应步骤中,淬灭反应所用的酸水溶液为盐酸水溶液、硫酸水溶液、硫酸氢钠水溶液、磷酸水溶液、柠檬酸水溶液中的一种或任意组合,优选为盐酸水溶液。
[0033]
进一步地,在所述分离步骤中,萃取所用的有机相选自与水不互溶的有机溶剂,如正丁醇、乙酸乙酯、乙酸异丙酯、甲苯、二甲苯、氯苯中的一种或任意组合,优选为正丁醇。
[0034]
进一步地,在所述第一混合步骤中,所述木糖溶液中的溶剂体积为溶质木糖体积的2~15倍,优选为10~12倍;所述酰化试剂溶液中,所用溶剂的体积为酰化试剂体积的1~15倍,优选为3倍。
[0035]
进一步地,在所述第一混合步骤中,所述木糖溶液、所述酰化试剂溶液和所述碱性溶液中溶质的摩尔比为1:(1.0~2.5):(1.0~2.0),优选为1:1.2:1.2。
[0036]
进一步地,在所述取代反应步骤中,反应温度为80~180℃,优选为120~150℃,反应时间为30~300秒。
[0037]
进一步地,在所述还原反应步骤中,所述木糖溶液中木糖与所述还原剂的摩尔比为1:(1.0~5),反应温度为20~80℃,反应时间为30~300秒。
[0038]
进一步地,在所述还原反应步骤中,所选还原剂的不同,木糖与还原剂选用不同的摩尔比值;当所述还原剂为硼氢化钠或硼氢化钾时,木糖的摩尔比与所述还原剂的摩尔比为1:(1.0~1.5);当所述还原剂为三乙酰氧基硼氢化钠时,木糖的摩尔比与所述还原剂的摩尔比为1:(3.0~5.0);反应温度为30~50℃,反应时间为30~300秒。
[0039]
进一步地,所述第一混合器、所述第二混合器和所述第三混合器均为微通道反应器、y型混合器、t型混合器或三通混合器。
[0040]
进一步地,所述第一反应器和所述第二反应器均为微通道反应器、管道反应器、碳化硅集束式反应器或折流板反应器。更加优选地,所述第一反应器为微通道反应器,即取代反应优选采用微通道反应器;所述第二反应器为折流板反应器,即还原反应优选折流板反应器。
[0041]
进一步地,所述油水连续分离器为油水分离器;或者,所述油水连续分离系统包括蒸馏器和萃取器。
[0042]
本发明的目的之二采用如下技术方案实现:
[0043]
一种连续流制备羟丙基吡喃三醇的合成系统,用于目的之一所述的合成方法,包括依次串联的进料系统、第一混合器、第一反应器、第二混合器、第二反应器、第三混合器和油水连续分离器;所述进料系统包括木糖溶液进料系统、酰化试剂溶液进料系统以及碱性溶液进料系统,所述木糖溶液进料系统、所述酰化试剂溶液进料系统以及所述碱性溶液进料系统分别与所述第一混合器连接。
[0044]
进一步地,所述油水连续分离器为油水分离器;或者,所述油水连续分离系统包括蒸馏器和萃取器。
[0045]
进一步地,所述第一混合器、所述第二混合器和所述第三混合器均为微通道反应器、y型混合器、t型混合器或三通混合器。
[0046]
进一步地,所述第一反应器和所述第二反应器均为微通道反应器、管道反应器、碳化硅集束式反应器或折流板反应器;更加优选地,所述第一反应器为微通道反应器,即取代反应优选采用微通道反应器;所述第二反应器为折流板反应器,即还原反应优选折流板反应器。
[0047]
相比现有技术,本发明的有益效果在于:
[0048]
(1)本发明所提供的连续流制备羟丙基吡喃三醇的合成方法,是一种连续流一锅法,将木糖、乙酰化试剂以及碱液分别泵入混合器和反应器中,保留一定的时间后,再泵入还原剂,反应一定时间后,用有机溶剂连续萃取和连续分液,可得羟丙基吡喃三醇的溶液,脱溶后即得羟丙基吡喃三醇。该方法反应时间短,操作简便,单体纯度高,自动化程度高,试剂廉价易得,具有良好的工业应用前景。
[0049]
(2)本发明所提供的连续流制备羟丙基吡喃三醇的合成方法,使用连续流工艺制
备,所得产品(式i化合物)纯度好,焦糖等杂质少,刺激性气味少,透明度高,异构体比例少,单一构型占比高。并且,本发明使用连续流反应时间短,安全性高,产能大,收率高,可降低环保污染。
[0050]
(3)本发明所提供的连续流制备羟丙基吡喃三醇的合成系统,构建简易,用于羟丙基吡喃三醇的连续流制备,自动化程度高,操作简便,具有良好的工业应用前景。
附图说明
[0051]
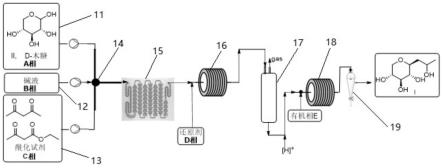
图1为本发明实施例所提供的连续流制备羟丙基吡喃三醇的合成方法的流程图;
[0052]
图2为本发明实施例2制备的羟基吡喃三醇的hplc图谱;
[0053]
图3为本发明实施例2和对比例1制备的羟基吡喃三醇的实物比对图;
[0054]
图4为对比例1制备的羟基吡喃三醇的hplc图谱。
[0055]
图1中:11、木糖溶液进料系统;12、碱性溶液进料系统;13、酰化试剂溶液进料系统;14、第一混合器;15、第一反应器;16、第二混合器;17、第二反应器;18、第三混合器;19、油水连续分离器。
具体实施方式
[0056]
下面,结合具体实施方式,对本发明做进一步描述,需要说明的是,在不相冲突的前提下,以下描述的各实施例之间或各技术特征之间可以任意组合形成新的实施例。
[0057]
本发明实施例提供了一种连续流制备羟丙基吡喃三醇的合成方法,反应流程如下:将式ii化合物d-木糖溶解于水或有机溶剂中,在碱液存在下,与乙酰化试剂(乙酰丙酮或乙酰乙酸乙酯)在微反应器中混合反应,所得中间体无需分离,接着直接于第二反应器中与还原试剂进行还原反应,然后经过淬灭后连续分液,脱溶后可得高浓度产品式i化合物,反应路线如下:
[0058][0059]
实施例1
[0060]
如图1所示,一种连续流制备羟丙基吡喃三醇的合成系统,包括依次串联的进料系统、第一混合器14、第一反应器15、第二混合器16、第二反应器17、第三混合器18和油水连续分离器19;进料系统包括木糖溶液进料系统11、酰化试剂溶液进料系统13以及碱性溶液进料系统12,木糖溶液进料系统11、酰化试剂溶液进料系统13以及碱性溶液进料系统12分别与第一混合器14连接。
[0061]
作为优选的实施方式,油水连续分离器19为油水分离器;或者,油水连续分离系统包括蒸馏器和萃取器。
[0062]
作为优选的实施方式,第一混合器14、第二混合器16和第三混合器18均为微通道反应器、y型混合器、t型混合器或三通混合器。
[0063]
进一步地,第一反应器15和第二反应器17均为微通道反应器、管道反应器、碳化硅集束式反应器或折流板反应器;更加优选地,第一反应器15为微通道反应器,即取代反应优
选采用微通道反应器;第二反应器17为折流板反应器,即还原反应优选折流板反应器。
[0064]
实施例2
[0065]
如图1所示,一种连续流制备羟丙基吡喃三醇的合成方法,包括以下步骤:将3.18kgd-木糖(式ii化合物)溶于32l水中,搅拌15分钟,d-木糖完全溶解,制备反应液a相。将2.69kg碳酸钠溶解于8l水中,制备反应液b相。将2.53kg乙酰丙酮溶解于7.6l正丁醇中,制备反应液c相。分别用柱塞泵设定a相流速为150ml/min;b相流速40ml/min;c相流速44ml/min;以设定流速恒定泵入第一混合器14中,然后进入第一反应器15(微通道反应器)中,反应温度设定125℃,保留时间为60秒。
[0066]
将1kgnabh4悬浮于4.5l水中,通过浆料泵以19ml/min的流速泵入第二混合器16中,设定第二混合器16的温度为50℃,保留时间为120秒,然后进入第二反应器17(折流板反应器)中,反应温度为50℃,保留时间为4分钟。
[0067]
将10%的盐酸以77ml/min的流速泵入带有构件的第三混合器18中,同时将正丙醇以220ml/min的流速泵入第三混合器18,混合后进入油水分离器进行分液。取有机相进行浓缩脱溶,可得目标化合物羟丙基吡喃三醇(式i所示),共得到产品3.53kg,为透明油状物,纯度大于99%,收率:87%,异构体比例小于1%。
[0068]
如图2所示,为实施例2制备的羟基吡喃三醇的hplc图谱,相对保留时间rt=2.261min为1,4-丁二醇,rt=4.881min为d-木糖,rt=5.788min为所需目标构型产品。
[0069]
实施例3
[0070]
如图1所示,一种连续流制备羟丙基吡喃三醇的合成方法,包括以下步骤:
[0071]
将3kgd-木糖(式ii化合物)溶于28l水中,搅拌15分钟,d-木糖完全溶解,制备反应液a相。将3.31kg碳酸钾溶解于10l水中,制备反应液b相。将3.12kg乙酰乙酸乙酯溶解于8.3l正丁醇中,制备反应液c相。分别用柱塞泵设定a相流速为150ml/min;b相流速40ml/min;c相流速44ml/min;以设定流速恒定泵入第一混合器14中,然后进入第一反应器15(微通道反应器),反应温度设定125℃,保留时间为40秒。
[0072]
将0.98kgnabh4悬浮于4.5l水中,通过浆料泵以19ml/min的流速泵入第二混合器16中,设定第二混合器16的温度为50℃,保留时间为120秒,然后进入第二反应器17(折流板反应器)中,反应温度为50℃,保留时间为4分钟。
[0073]
将10%的盐酸以70ml/min的流速泵入带有构件的第三混合器18,同时将正丙醇以200ml/min的流速泵入第三混合器18,混合后进入油水分离器进行分液。取有机相进行浓缩脱溶,可得目标化合物羟丙基吡喃三醇(式i所示),共得到产品3.27kg,为透明油状物,纯度大于99%,收率:85%,异构体比例小于1%。
[0074]
实施例4
[0075]
如图1所示,一种连续流制备羟丙基吡喃三醇的合成方法,包括以下步骤:
[0076]
将9kgd-木糖(式ii化合物)溶于84l乙醇中,搅拌30分钟,d-木糖完全溶解,制备反应液a相。将2.88kg氢氧化钠溶解于30l水中,制备反应液b相。将7.2kg乙酰丙酮溶解于25l乙醇中,制备反应液c相。分别用柱塞泵设定a相流速为450ml/min;b相流速120ml/min;c相流速132ml/min;以设定流速恒定泵入第一混合器14中,然后进入第一反应器15(微通道反应器),反应温度设定125℃,保留时间为40秒。
[0077]
将3.2kgnabh4悬浮于15l水中,通过浆料泵以60ml/min的流速泵入第二混合器16
中,设定第二混合器16的温度为50℃,保留时间为110秒,然后进入第二反应器17(折流板反应器)中,反应温度为50℃,保留时间为4分钟。
[0078]
将10%的盐酸以200ml/min的流速泵入带有构件的第三混合器18,反应完毕,将溶液减压蒸馏除去乙醇,然后用正丙醇萃取三次,合并有机相进行减压浓缩脱溶,可得目标化合物羟丙基吡喃三醇(式i所示),共得到产品10kg,为透明油状物,纯度大于99%,收率:87%,异构体比例小于1%。
[0079]
实施例5
[0080]
如图1所示,一种连续流制备羟丙基吡喃三醇的合成方法,包括以下步骤:
[0081]
将1.8kgd-木糖(式ii化合物)溶于16.8l乙醇中,搅拌30分钟,d-木糖完全溶解,制备反应液a相。将576g氢氧化钠溶解于6l水中,制备反应液b相。将1.44kg乙酰乙酸乙酯溶解于5l乙醇中,制备反应液c相。分别用柱塞泵设定a相流速为450ml/min;b相流速120ml/min;c相流速132ml/min;以设定流速恒定泵入第一混合器14中,然后进入第一反应器15,反应温度设定125℃,保留时间为40秒。
[0082]
将3.5kgnabh(oac)3悬浮于3l水中,通过浆料泵以60ml/min的流速泵入第二混合器16中,设定第二混合器16的温度为50℃,保留时间为110秒,然后进入第二反应器17(折流板反应器)中,反应温度为50℃,保留时间为4分钟。
[0083]
将10%的盐酸以200ml/min的流速泵入带有构件的第三混合器18,反应完毕,将溶液减压蒸馏除去乙醇,然后用正丙醇萃取三次,合并有机相进行减压浓缩脱溶,可得目标化合物羟丙基吡喃三醇(式i所示),共得到产品1.9kg,为透明油状物,纯度大于99%,收率:83%,异构体比例小于1%。
[0084]
对比例1
[0085]
按照专利202110383018.4一种一锅法合成玻色因的方法的实施例1进行操作,具体步骤如下:在1000ml的反应瓶中依次加入乙醇500g、木糖30g(0.2mol)、碳酸氢钠0.17g(2mmol)、乙酰乙酸乙酯26g(0.2mol),搅拌升温至60℃,继续反应1h,反应液用tlc检测至木糖基本消失(gf254硅胶板,展开剂为二氯甲烷:甲醇=4:1,20%硫酸乙醇溶液显色)。然后在0℃时逐滴加入硼氢化钠11.3g(0.3mol),搅拌均匀后在50℃下反应24h后,缓慢滴加2n盐酸,调ph至中性,再用乙酸乙酯萃取分液,收集有机层,旋干,得到玻色因,为焦糖色油状物,纯度为12.7%,收率为77%,异构体比例为46.8%。如图4所示,为对比例1制备的的羟基吡喃三醇的hplc图谱,相对保留时间rt=5.793min为所需目标构型产品。
[0086]
如图3所示,为本发明实施例2和对比例1所得羟丙基吡喃三醇的实物图,图中可得,对比例1得到的玻色因的产品颜色为焦糖色,并且醋酸味很重,这主要是因为对比例1的反应时间较长。相反,本发明实施例2采用了连续流合成方法,得到的玻色因玻色因颜色为无色透明溶液,醋酸味道很淡,几乎没有酸味,这对于化妆品来说是非常重要的改进。实施例3-5的产品实物图与实施例1类似,再此不再赘述。
[0087]
本发明实施例所提供的连续流制备羟丙基吡喃三醇的合成方法,反应时间短,操作简便,自动化程度高,试剂廉价易得,所得产品(式i化合物)纯度好,焦糖等杂质少,刺激性气味少,透明度高,异构体比例少,单一构型占比高。同时制备方法安全性高,产能大,收率高,可降低环保污染,具有良好的工业应用前景。
[0088]
上述实施方式仅为本发明的优选实施方式,不能以此来限定本发明保护的范围,
本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
