一种活化过硫酸盐和o2协同降解全氟化合物的电化学方法
技术领域
1.本发明属于水处理技术领域,具体涉及一种活化过硫酸盐和o2协同降解全氟化合物的电化学方法。
背景技术:
2.全氟类化合物(pfcs)是人工合成的有机物,其所有烷基链上的氢原子都被氟原子取代,其分子结构既有全氟的疏水性烷基链又有亲水的官能团,化学性质极为稳定,不可燃,且与大部分酸、碱、强氧化剂和强还原剂不发生反应,而被广泛应用于制造灭火剂、产品包装、皮革、纸张等方面。它广泛且持久地存在于自然界中,难以降解,污染水体环境,且其毒性富集在人体和动物的血液、心脏、肝脏等器官中,对环境和人类健康造成一定的威胁。
3.pfcs之所以化学性质稳定,是因为c-f键化学键能高(116kcal/mol),还原电势高(f e-=f-,e0=3.6v),氟原子的2s和2p轨道与碳原子相应轨道之间的完美重叠,使得碳链被性质稳定的氟原子紧密包裹,而每个氟原子有3对孤电子对,有效屏蔽化学物质对c-c单键的亲核攻击。由于pfcs的高稳定性,其在环境中可以持久地存在且广泛分布于各种环境介质中,其中全氟辛酸(pfoa)和全氟辛烷磺酸(pfos)被检出频率最高,是目前最受关注的典型pfcs。有研究发现,pfos和长碳链的全氟羧酸(c9-c12)比短碳链的pfcs呈现出更强的生物累积性,且能够随着食物链显著放大。为了实现对pfcs的快速降解,高级氧化/还原技术应用广泛。其中,基于电化学的高级氧化/还原技术具有反应条件温和、操作简单、环境友好且易于控制等优点,在去除废水中有机污染物方面有着广泛应用。电化学方法去除pfcs的过程是:首先,pfcs(c
nf2n 1
coo-/cnf
2n 1
so
3-)必须失去一个电子形成pfcs自由基(c
nf2n 1
coo
·
/cnf
2n 1
so3·
),继而脱羧或脱磺酸基形成全氟烷基自由基(c
nf2n 1
·
);随后,该自由基容易与
﹒
oh形成不稳定中间体c
nf2n 1
oh;然后,再进一步反应形成新的短链pfcs或短链pfcs自由基;最后,通过循环氧化,将长链逐渐降解至短链pfcs,直至被完全矿化成co2和f-。
4.目前,基于水体中全氟化合物电化学去除技术有电化学氧化、电絮凝、以及电吸附技术等。电化学虽然能高效且深度地降解pfcs,但是仍存在能耗高、电极材料不稳定、pfoa/pfos在阴阳极之间传质阻力大等缺点。因此本工作提出一种在铁镍双金属共掺杂碳气凝胶阴极材料表面同时产生硫酸根自由基(so4·-)和羟基自由基(
·
oh),协同降解和矿化pfcs的体系。在电催化体系中,发生两电子氧还原反应原位产生h2o2,然后h2o2在阴极材料表面丰富的金属活性位点上被催化产生﹒oh,同时金属活性位点也能够活化pms产生so4·-。已有研究表明,过渡金属铁和镍可活化过硫酸盐产生硫酸根自由基,同时镍的掺杂有利于提高材料的两电子氧还原选择性,从而产生过氧化氢,并进一步转化为﹒oh。因此,铁镍共掺杂碳气凝胶有望实现在阴极表面同时产生硫酸根自由基和羟基自由基的目的。实验结果表明,该体系对短碳链的全氟羧酸中间产物具有明显增强的氧化矿化能力,且通过调节fe、ni前驱体的比例,能够产生不同比例的so4·-和
·
oh,增强对特定碳链长度全氟羧酸的降解矿化能力,达到高效去除pfcs的目的。
技术实现要素:
5.因此,本发明要解决现有技术中存在的缺陷,从而提供一种活化过硫酸盐和o2协同降解全氟化合物的电化学方法。
6.为此,本发明提供了以下技术方案,
7.本发明提供了一种活化过硫酸盐和o2协同降解全氟化合物的电化学方法,包括以下步骤,
8.采用二电极直流电源,以20~30ma
·
cm-2
的恒电流密度,将活化后的“fe-ni-c”气凝胶电极作为阴极,将石墨片作为阳极,使全氟化合物在0.04m的过氧单磺酸钾电解质溶液中进行降解,以100-200ml/min的速率持续通氧气,始终维持在氧饱和的状态,降解时间为4h,反应温度为20-25℃,ph范围为2-5。
9.可选的,所述的全氟辛酸浓度为50ppm。
10.可选的,所述活化后的feni-ca的制备方法,包括以下步骤,
11.s1:将间二苯酚、甲醛、碳酸钠加入水中,混合搅拌均匀,然后加入fe源和ni源搅拌均匀后密封,得到溶液;
12.s2:将步骤s1得到的溶液置于20-30℃下,恒温24-30h,然后置于40-50℃下,恒温24-30h,最后置于80-90℃下,恒温24-30h,得到“fe-ni-c”气凝胶前驱体;
13.s3:将步骤s2得到的“fe-ni-c”气凝胶前驱体在丙酮溶液中浸泡3d,浸泡后在室温下干燥2-3d,得到块状“fe-ni-c”气凝胶阴极材料。
14.可选的,所述活化后的feni-ca的制备方法,还包括以下步骤,
15.s4:将步骤s3得到的块状“fe-ni-c”气凝胶阴极材料置于管式炉中,在n2的氛围下,进行焙烧,然后冷却至室温,得到“fe-ni-c”气凝胶电极;
16.s5:将步骤s4得到的“fe-ni-c”气凝胶电极置于co2管式炉中,在co2氛围下,进行焙烧,然后冷却至室温,得到活化后的feni-ca。
17.可选的,步骤s4中n2的流速为200-300ml/min;
18.步骤s4中所述焙烧是以2.5-5℃/min的速率升温至850-950℃;
19.步骤s4中所述焙烧时间为2-3h;
20.可选的,步骤s5中co2的流速为100-200ml/min;
21.步骤s5中所述焙烧是以2.5-5℃/min的速率升温至650-750℃;
22.步骤s5中所述焙烧时间为1-2h。
23.可选的,所述的间苯二酚:水:甲醛:碳酸钠的摩尔比为1:16-18:2-3:0.0008-0.001。
24.可选的,所述的间苯二酚:水:甲醛:碳酸钠的最优摩尔比为1:17.5:2:0.0008。
25.可选的,所述fe源为乙酰丙酮铁;
26.所述ni源为乙酰丙酮镍。
27.可选的,所述fe源和ni源的加入量分别占原料的总质量的2%-6%。
28.本发明提供的技术方案,具有如下优点,
29.1.本发明通过一步合成法原位形成掺杂了铁和镍的双金属碳气凝胶,以ca为基底,具有较高的电化学活性和较大的比表面积,能够提供丰富的反应活性位点,不仅能够在电极表面原位发生2电子氧还原反应(orr),产生h2o2,同时电极表面丰富的金属活性位点在
催化h2o2产生
·
oh的同时也能够活化pms产生so4·-。
30.2.本发明通过优化调节铁和镍的质量比分别得到fec、fe3ni1c、fenic、fe1ni3c、nic这五种材料,相比单独的fec和nic,fe、ni共掺杂时有利于活性位点的暴露,减少传质阻力,增加碳缺陷的形成;铁和镍共同存在时有利于提高碳气凝胶材料的稳定性,能够使碳基底负载大量稳定的金属活性位点。其中,fe、ni质量比为1:1时,fenic具有最高的比表面积(727.55m2·
g-1
)、适中的亲疏水性(126
°
)以及大量的缺陷碳(68.64%),具有最佳的电催化活性,可作为阴极用于电fenton过程,并活化过氧单磺酸盐(pms)同时产生羟基自由基和硫酸根自由基,在阴极表面降解全氟化合物。
31.3.本发明与传统的降解pfcs方法相比,如吸附法、膜分离法、生物处理法以及光催化法等等,本发明的fe、ni双金属共掺杂电极材料能够实现在阴极同步产生硫酸根自由基和羟基自由基协同降解全氟羧酸,避免了在阴阳极之间的传质问题,提高降解效率,具有低能耗、操作简便等特点。
32.4.本发明制备所得的fenic材料不仅在阴极有着良好的电fenton活性,同时利用了其表面所负载的丰富的金属活性位点活化pms高效产硫酸根自由基,在酸性条件下,实现对在水环境中具有持久性和生物累积性的pfcs高效降解矿化。
附图说明
33.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
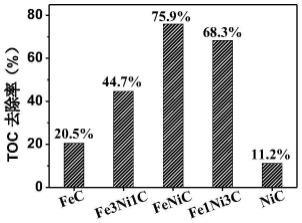
34.图1为不同比例feni-ca阴极4h降解pfoa的toc去除率;
35.图2为不同比例feni-ca阴极4h降解pfoa的脱氟率;
36.图3为不同比例feni-ca阴极产生硫酸根自由基和羟基自由基的epr图谱。
具体实施方式
37.提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
38.实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
39.实施例1
40.本实施例中“feni-ca”或“fec”以及“nic”气凝胶的具体制备方法如下:
41.(1)称取适量的间苯二酚、甲醛、水、碳酸钠,间苯二酚:甲醛:水:碳酸钠的摩尔比为1:17:2:0.0008混合均匀,将前驱体溶液分成五等份,然后称取不同比例的乙酰丙酮铁、乙酰丙酮镍,使m
fe
/mc=2-3%,m
(fe ni)
/mc=2-5%,m
(fe ni)
/mc=2-6%,m
(fe ni)
/mc=3-5%,m
ni
/mc=2-3%,加入至上述混合溶液中分散均匀,然后注入玻璃容器中密封。
42.(2)将密封的玻璃容器放入烘箱,设置20-30℃条件下,恒温24-30小时,40-50℃恒温24-30小时,最后于80-90℃条件下恒温24-30小时以发生聚合反应,形成块状湿凝胶。将制备成形的湿凝胶浸泡在丙酮溶液中,浸泡三天,每天更换,来置换湿碳气凝胶网格结构中的水分,浸泡完成后于室温下干燥2-3天,最终得到完整的块状fec、nic及不同比例feni-ca碳气凝胶阴极材料。
43.(3)将上述系列碳气凝胶材料放入管式炉中焙烧、活化。在流速为200-300ml/min的氮气气氛中,以2.5-5℃/min的速率升温至850-950℃的条件下焙烧,并在该温度下保持2-3h,之后以同样的速率降至室温,初步得到焙烧后的fec、nic及不同比例feni-ca碳气凝胶材料,随后再将其置于管式炉中,进一步在流速为100-200ml/min的co2气氛中,以2.5-5℃/min的速率升温至650-750℃的条件下,扩孔活化,在该温度下保持1-2h,然后以相同的速率降至室温,最终得到经n2焙烧、co2活化的完整的fec、nic及不同比例feni-ca碳气凝胶阴极材料。
44.实施例2
45.分别以不同比例铁镍碳气凝胶为阴极,以石墨片为阳极,对阴极原位产生过氧化氢的产量进行研究。在单室反应池中进行双电极体系电催化orr过程,每隔0.5h取1ml样品,加入同体积6g/l-1
硫酸氧钛溶液中,形成稳定的橙色络合物。通过紫外-可见分光光度计对h2o2的量进行测定,波长为λ=508nm,根据吸光度与浓度的关系得到不同样品中过氧化氢的浓度。不同比例铁镍碳气凝胶作为阴极时体系产生h2o2的能力由高到低依次为fe1ni3c、fenic、fe3ni1c、fec和nic,且在反应4h后,体系中h2o2的浓度分别为62.3ppm、43.9ppm、38.5ppm、14.7ppm以及8.5ppm。进一步使用捕获剂dmpo与so4·-以及
·
oh络合,并通过电子顺磁共振仪检测两种自由基的信号,如图3所示,fec、fe3ni1c、fenic和fe1ni3c阴极均能同时产生so4·-和
·
oh,nic阴极能观测到较微弱的
·
oh信号。其中fenic阴极产生两种自由基的能力最强,比例最为接近。
46.实施例3
47.将实施例1中fec、nic及不同比例feni-ca阴极材料分别应用到电催化反应体系中,评估在该系列阴极材料表面原位降解全氟羧酸的能力,选择全氟辛酸(pfoa)为代表性的全氟化合物。将其作为电催化反应体系中的阴极,其中阳极为石墨片电极,电极面积均为6cm2。使用二电极恒电位仪作为直流电源,施加20-30ma/cm-2
的恒定电流密度,电压变化在2-3.5v。pfoa(30ml,50ppm)在0.04m的过氧单磺酸钾溶液为支持电解质中进行降解反应。通电前先以100-200ml/min的速率持续通氧气10-15min,降解过程中始终维持在氧饱和的状态,降解时间为4h,反应温度为20-25℃,ph范围为2-5。由图1、2可知,将fec、fe3ni1c,fenic、fe1ni3c、nic分别作为电催化阴极时,降解4h后的toc去除率分别为20.5%、44.7%、75.9%、68.3%以及11.2%,脱氟率为10.8%、50.1%、63.3%、54.3%和5.3%。可见,其中fenic作为电催化阴极材料时降解效率最好。进一步评估fenic的稳定性,循环使用20h后,toc去除率降低了5.6%,脱氟率降低了6.7%。
48.实施例4
49.评估fec、nic及不同比例feni-ca作为电化学阴极材料在表面原位降解全氟类化合物的能力,选择全氟辛烷磺酸(pfos)为代表性的全氟类化合物。将其作为电催化反应体系中的阴极,其中阳极为石墨片,电极面积均为6cm2。使用二电极恒电位仪作为直流电源,
施加20-30ma/cm-2
的恒定电流密度,电压变化在2-3.5v。pfos(30ml,50ppm)在0.04m的过氧单磺酸钾溶液为支持电解质中进行降解反应,通电前先以100-200ml/min的速率持续通氧气10-15min,降解过程中始终维持在氧饱和的状态,降解时间为4h,反应温度为20-25℃,ph范围为2-5。fec、fe3ni1c,fenic、fe1ni3c、nic分别作为电催化阴极时,降解4h后,对pfos的降解率分别为21.5%、58%、81.6%、65.3%以及16.8%,其中fenic阴极材料对pfos的降解效率最高。
50.显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。