一种检测肺炎支原体及其23SrRNA基因耐药突变的生物信息分析方法与流程
- 国知局
- 2024-07-12 10:33:53
本发明属生物医药领域,具体涉及一种检测肺炎支原体大环内酯类药物耐药位点的生物信息分析方法。
背景技术:
1、肺炎支原体(mycoplasma pneumoniae,mp)是一种细菌,属于支原体目中的一种。它是人类呼吸道常见的致病菌之一,引起的疾病主要包括上呼吸道感染、肺炎、支气管炎等。肺炎支原体肺炎(mpp)是我国5岁及以上儿童最主要的社区获得性肺炎,占住院儿童社区获得性肺炎的10%至40%;临床上用于治疗肺炎支原体感染的首选抗菌药物是大环内酯类药物,但是,随着该药物在临床上的广泛应用,其耐药现象也不断出现,并呈逐渐增加趋势。除此之外,由于肺炎支原体缺乏细胞壁结构,因此对青霉素类、头孢类抗生素等作用于细胞壁的抗生素固有耐药。
2、目前,已有大量研究表明,肺炎支原体耐大环内酯类药物的机制主要为基因突变,其中23s rrna基因的突变是其对大环内酯类药物耐药的主要原因之一;大环内酯类药物的结合位点在23s rrna结构域,23s rrna结构域ⅱ区和ⅴ区基因位点变异会降低抗菌药物和核糖体之间的亲和力,从而使肺炎支原体产生耐药性。已发现的变异位点包括2063、2064、2067和2617,其中a2063g阳性表现为对14-环大环内酯类耐药,a2064g阳性表现为对14-和16-环大环内酯类耐药,c2617g阳性表现为对14-和15-环大环内酯类抗菌药物耐药,a2067g阳性表现为对交沙霉素耐药。
3、肺炎支原体及其耐药基因的检测方法虽然多种多样,但是基于分离培养的肺炎支原体检测及药物敏感性试验无疑是“金标准”,该法虽然可靠,但存在肺炎支原体生长缓慢、培养周期长、技术要求高、分离费用昂贵,分离培养阳性率低等诸多缺点,以致临床应用困难。除此之外,常用的肺炎支原体耐药检测技术还有以pcr为代表的分子生物学技术,比如毛细管电泳、数字pcr和荧光定量pcr等,该方法的关键是针对耐药位点设计特异性引物探针,但常常由于引物设计不合理、特异性不够强等问题,导致无法实现准确、快速、灵敏的检测,以致实际应用受到很大限制。
技术实现思路
1、为了解决上述问题,本技术提供了一种检测肺炎支原体及其23s rrna基因耐药突变的生物信息分析方法,不需要进行细菌分离培养,避免了传统方法中可能出现的细菌培养失败的情况,同时能够在较短时间内对大量数据进行分析,加快耐药性基因突变的检测速度。
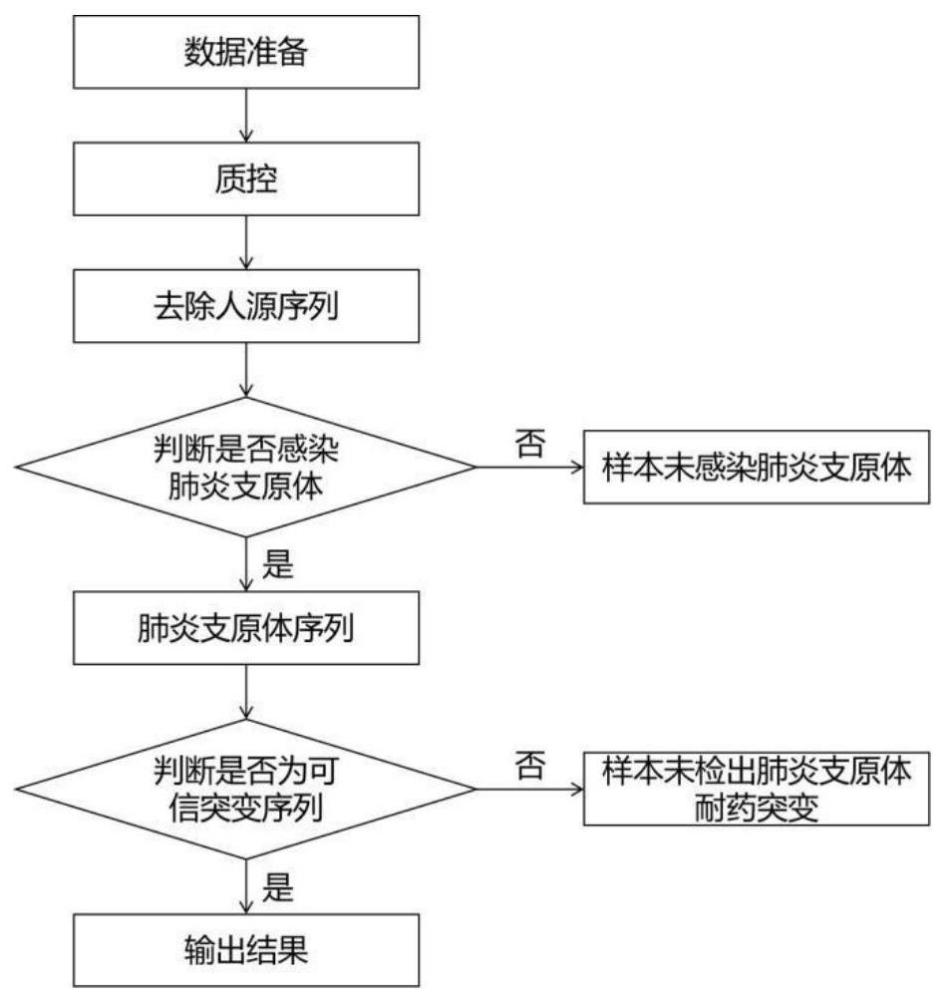
2、本技术提供的方法具体包括如下步骤:
3、1.数据准备,需要准备的数据包括ncbi数据库标注的肺炎支原体参考基因组、23srrna基因的序列、hg19版本的人类参考基因组以及转录组序列、炎黄一号人类基因组、t2t-chm13人类参考基因组序列、病原宏基因组(mngs)临床样本以及阴控样本的二代测序数据。
4、2.病原宏基因组(mngs)临床样本以及阴控样本的二代测序数据的质控,具体质控条件如下:
5、(1)去除接头(adapter)序列,在高通量测序过程中,当插入片段的长度小于测序的读长时,接头序列就会出现在测序结果中引起污染,因此质控的时候要将其进行切除;
6、(2)去除首尾的碱基质量值低于30的碱基;
7、(3)去除read中含有n碱基的序列;
8、(4)去除单一碱基占比超过80%或者两种碱基占比超过90%的序列;
9、(5)去除重复序列;
10、(6)去除低复杂度序列,或者叫简单重复序列,低复杂度序列往往是测序错误的高发区,质控的时候要加以去除。
11、在本技术中,判断一个样本在质量控制之后是否可以使用要同时满足以下条件:
12、(1)q30碱基数据量占比大于80%;
13、(2)接头污染比例不超过1%;
14、(3)有效序列长度不小于45bp;
15、(4)数据的有效数据量应大于70%。
16、3.构建人源序列数据库,本技术构建了一个全面且和中国人种适配的人源序列数据库,除了常规的人类参考基因组与转录组之外,还新增了专门针对中国人种的炎黄一号基因组序列以及人类基因组序列无间隙版本t2t-chm13;将质控后的数据与人源序列数据库进行比对,去除比对上的序列,剩下的即为高质量非人源序列。
17、4.采用bwa软件对肺炎支原体参考基因组序列建立索引,并把步骤3中的高质量非人源序列往参考基因组序列上进行比对,并采用samtools软件对比对结果进行排序、建立索引等处理。
18、5.制定比对序列筛选条件,从步骤4中的比对结果中提取符合下述条件的比对序列并保存成fastq格式的文件用来寻找耐药突变。
19、(1)比对到肺炎支原体参考基因组上的平均测序质量值大于等于30且比对质量值(mapq值)大于等于30的read数目n大于等于10条;
20、(2)只比对到肺炎支原体参考基因组上的平均测序质量值大于等于30且比对质量值(mapq值)大于等于30的read数目大于等于2条,也即唯一比对的read(uniquely mappedread)数目大于等于2条;
21、(3)临床样本以及阴控样本的均一化之后的比对read数之比大于等于5,也即foldchange大于等于5;如果阴控样本中没有满足条件的read检出,则foldchange为正无穷。
22、样本均一化方法如下:均一化后的序列数=(20*n*0.000001)/样本大小(单位m)
23、则:foldchange=临床样本均一化后序列数/阴控样本均一化后序列数
24、6.采用bwa软件对23s rrna基因序列建立索引,并把步骤5中的提取到的fastq格式的序列往23s rrna基因组上进行比对,并采用samtools软件对比对结果进行排序、建立索引等处理,处理之后生成相应的bam格式文件。
25、7.采用bam-readcount软件从步骤6中生成的bam文件里统计23s rrna基因2063、2064、2067以及2617四个位点的a、c、g、t四种碱基以及n的个数,其中n表示测序时不确定是具体那种碱基,acgtn的数目之和为该位点的测序深度。
26、8.根据制定的筛选原则从步骤7中的碱基组成结果里筛选可信的突变结果,具体筛选原则如下:
27、(1)发生突变的位点在单条链(正链或者负链)的占比不得超过90%;
28、(2)突变频率大于等于50%且突变read数目大于等于2;
29、(3)突变频率大于等于5%且小于50%且突变read数目大于等于2且突变位点总深度大于等于10;
30、在满足(1)的基础上,条件(2)和(3)中满足任一条件即为可信的突变结果。
31、与现有技术相比,本技术具有以下有益效果:
32、(1)本发明方法具有高度敏感性、强大的特异性和良好的重复性,且所需时间较短,适用于肺炎支原体及其23s rrna耐药位点的检测,这一方法在临床诊疗中具有广泛的应用前景。
33、(2)在肺炎支原体鉴定方面,与传统的分离培养法相比,本发明提供的方法更为高效快速且成本更低;在耐药检测方面,该方法也有别于传统的药敏试验以及特异性pcr引物设计方法;除此之外,本发明还自建了一套突变分析方法,该方法从耐药位点的碱基组成入手,与现成的变异检测软件相比极大地提高了检测的灵敏度。
本文地址:https://www.jishuxx.com/zhuanli/20240614/87744.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表