1.本发明涉及天然药用化合物技术领域,具体涉及一种新化合物、从双蝴蝶中提取分离该化合物的方法及该化合物抗炎的制药用途。
背景技术:
2.双蝴蝶tripterospermumchinense(migo)h.smith为龙胆科双蝴蝶属植物双蝴蝶的全草,又名肺形草、黄金线。它主产于浙江、安徽、江西等地,具有清热解毒、止咳止血等功效,适用于治疗支气管炎、肺痨咯血、肺炎、肺脓肿、肾炎、淋证,也可外用于疗疮疖肿、乳腺炎及外伤出血。现代研究表明,双蝴蝶植物中含有呫吨酮类、环烯醚萜类及黄酮类等物质,药理实验研究发现双蝴蝶具有抗炎抗病毒、抗高血压、抑制中枢神经等作用。
3.在中国专利公开文献中,由吉首大学申请的公开号为cn103142684a的发明专利申请公开了一种峨眉双蝴蝶提取物及其制备方法,制备方法具有工艺步骤简单、产品纯度高等优点,峨眉双蝴蝶提取物广泛应用于原料药、医药中间体和保健食品领域。公开号为cn105454866a的发明专利申请公开了一种肺形草清肺止咳汤料及其生产方法,使得各种药物的药效产生协同作用,从而能够达到效果,长期使用具有清肺止咳、清热化痰、润肺平喘的功效。由江西中医药大学申请的公开号为cn111281921a发明专利申请公开了一种治疗感冒、肺炎、肠炎、肾炎的肺形草银翘解毒消炎组合物,发挥了中医药治疗肺炎并伴有肠炎和肾炎辩证论治、病证结合的特点与优势,提供了一种治疗感冒、肺炎、肠炎、肾炎的肺形草银翘解毒消炎组合物的制备方法及其在制备治疗感冒、肺炎、肠炎、肾炎药物中的应用,提供了肺形草银翘解毒消炎组合物新的制备工艺及其具体新用途和新功效。
4.目前,国内外学者对中药双蝴蝶的生物活性研究主要是提取物,对其所含化学成分的具体药理活性研究较少,多与其他药材配伍使用,成分复杂,其作用机制尚不明确,较少见有关双蝴蝶环烯醚萜苷成分抗炎的相关文献,且其环烯醚萜苷的结构具有较强的特异性,因此,从分离鉴定其中的单体化合物入手,找到其发挥各药效的有效成分,以便进行深入的研究与开发利用,是具有十分重要意义的。
技术实现要素:
5.针对上述现有技术中存在的问题和不足,本发明的目的在于从双蝴蝶中提取得到一种新化合物,同时提供其抗炎的制药用途。
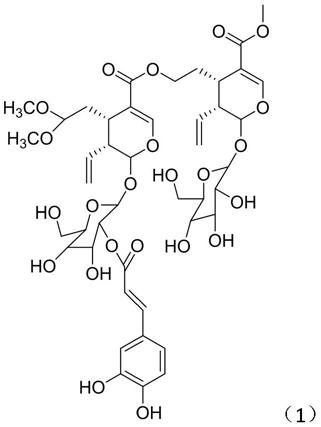
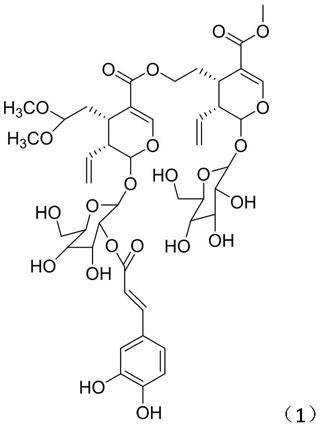
6.为实现本发明的上述目的,本发明从双蝴蝶中提取得到新化合物:化学式为 c
44
h
58
o
23
,名称为tripterospermumcin j,结构式如下式(1)所示:
[0007][0008]
从双蝴蝶中提取上述新化合物式(1)的方法,其步骤如下:取干燥的双蝴蝶全草,粉碎后,依次用95v/v%乙醇和60v/v%乙醇采用渗漉法提取浓缩后,用石油醚、乙酸乙酯依次进行萃取,得到的乙酸乙酯部位萃取物依次经过硅胶柱色谱、sephadex lh
‑
20和ods
‑
hplc分离纯化,得到新化合物。
[0009]
另外,本发明从酶活实验和分子对接两个方面开展新化合物的抗炎活性评价,新化合物对mif具有较好的抑制活性,其对mif的ic50为12.7μmol/l。提取分离的活性新化合物可以应用于制备抗炎的药品和保健品,优选作为mif的抑制剂。
附图说明
[0010]
图1为本发明新化合物的ir谱图;
[0011]
图2为本发明新化合物的uv谱图;
[0012]
图3为本发明新化合物的hr
‑
esi
‑
ms谱图;
[0013]
图4为本发明新化合物的1h
‑
nmr光谱图;
[0014]
图5为本发明新化合物的
13
c
‑
nmr光谱图;
[0015]
图6为本发明新化合物的核磁共振hmqc光谱图;
[0016]
图7为本发明新化合物的核磁共振hmbc光谱图;
[0017]
图8为本发明新化合物的核磁共振nosey光谱图;
[0018]
图9为本发明新化合物的核磁共振cosey光谱图;
[0019]
图10为本发明新化合物与靶蛋白mif的二维和三维效果图。
具体实施方式
[0020]
下面申请人将结合本发明的实施例及说明书附图,对本发明的技术方案进行清楚、完整的描述。
[0021]
实施例1
[0022]
化合物的提取制备
[0023]
步骤1、取干燥的双蝴蝶(tripterospermumchinense(migo)h.smith)(产地:浙江温州)的全草16.0kg,粉碎过20目筛后,依次用95v/v%乙醇(120l) 和60v/v%乙醇(120l)
采用渗漉法提取,合并浸出液,浓缩后得总浸膏6.5kg;
[0024]
步骤2、将步骤1得到的总浸膏用水混悬后,用石油醚、乙酸乙酯依次进行萃取,分别得到石油醚部位萃取物103.0g、乙酸乙酯部位萃取物231.0g;
[0025]
步骤3、将步骤2得到的乙酸乙酯部位萃取物(231.0g)采用硅胶柱色谱技术,以石油醚:乙酸乙酯:甲醇体系进行梯度洗脱,体积比依次为:1:0:0、10:1:0、 6:1:0、4:1:0、3:1:0、2:1:0、1:1:0、1:2:0、1:3:0、1:4:0、0:1:0、0:95:5、0:90:10,收集石油醚:乙酸乙酯:甲醇体积比0:95:5
‑
0:90:10的洗脱液,编号为fr.c,经减压浓缩至干,备用;
[0026]
步骤4、步骤3所得组分fr.c经硅胶柱色谱分离,以二氯甲烷
‑
甲醇(体积比依次为80:20、70:30、60:40)体系进行梯度洗脱,收集二氯甲烷
‑
甲醇体积比 60:40的洗脱液,编号为fr.ce,减压浓缩至干;再经sephadex lh
‑
20分离,以氯仿
‑
甲醇(体积比1:1)为洗脱系统,洗脱液用量为1.5个柱体积,收集其中第 0.8
‑
1.1柱体积部分的洗脱液,编号为fr.ce3,减压浓缩至干;最后用ods
‑
hplc 纯化,洗脱剂为meoh
‑
h2o,meoh:h2o体积比45:55,色谱柱为c
‑
18柱(5μm, 250mm
×
10mm),流速3.0ml/min,收集第25
‑
27分钟的洗脱液,减压浓缩干燥,得到新化合物10.0mg。
[0027]
实施例1所得新化合物结构鉴定:采用ir、uv、现代波谱技术如1h nmr 核磁谱、
13
c nmr核磁谱、二维核磁谱(hmqc、hmbc、nosey、cosey)、高分辨质谱(hr
‑
esi
‑
ms)对步骤4所得新化合物进行结构鉴定,结果见图1
‑
9;
[0028]
经鉴定,步骤4所得新化合物经hr
‑
esi
‑
ms给出准离子峰 977.32538[m na]
,结合1hnmr、
13
c nmr、二维核磁谱确定其分子式为 c
44
h
58
o
23
,名称为tripterospermumcinj,结构式如下式(1);其核磁共振谱图数据如表1所示。
[0029][0030]
表1:新化合物的1h(600mhz,cdcl3),
13
c(150mhz,cdcl3)和hmbc核磁数据
[0031]
[0032][0033]
为检测新化合物对macrophage migration inhibitory factor(mif)的抑制活性,现做以下酶活实验:
[0034]
mif酶活实验的实验原理为:在mif(巨噬细胞移动抑制因子)催化下,橙色底物l型多巴色素甲酯转化成无色的2,3
‑
吲哚二酸衍生物。这一颜色的改变可以在475nm波长下检
测到,吸光度值降得越低,说明酶的活性越强。当加入抑制剂后,抑制剂阻碍底物(l型多巴色素甲酯)与mif的结合,使底物不能转化成2,3
‑
吲哚二酸衍生物,导致吸收差值变小。
[0035]
步骤1、10mmol/l含0.5mmol/l edta钾磷酸缓冲液的配制:称取0.1061g 磷酸钾,加水溶解,定容至50ml,得到浓度为10mmol/l的磷酸钾缓冲液,然后称取0.0146g edta溶于上述磷酸钾缓冲液中,待溶解后,用ph计测混合后溶液的ph值,然后加入适量磷酸氢二钾溶液或磷酸调节ph至6.2,即得。以下实验操作提及该缓冲液简记为:10mmol/l磷酸钾缓冲液(0.5mm edta, ph=6.2)。
[0036]
步骤2、底物溶液的配制:称取1.486mg盐酸左旋多巴甲酯溶于 1.5ml10mmol/l磷酸钾缓冲液(0.5mm edta,ph=6.2),置于4℃保存。称取 2.567mg高碘酸钾溶于1.5ml纯水,置于4℃保存。使用前5min将等体积的盐酸左旋多巴甲酯溶液和高碘酸钾溶液混合制备成橙色的l
‑
多巴色素甲酯,冰上备用。
[0037]
步骤3、mif酶溶液的配制:将10μgmif酶用667μl10mmol/l磷酸钾缓冲液(0.5mm edta,ph=6.2)溶解,配成1200nmol/l的酶液,分装于ep管中,置于
‑
80℃的冰箱中冻存。使用前稀释至120nmol/l,冰上备用。
[0038]
步骤4、药物(新化合物和阳性药)的配制:
[0039]
新化合物溶液的配制:称取0.00977g实施例1步骤4所得新化合物,溶解于0.5mldmso,得浓度为20mmol/l母液。
[0040]
阳性药(iso
‑
1)溶液的配制:称取2mg iso
‑
1(巨噬细胞移动抑制因子拮抗剂),溶解于425μl dmso,得浓度为20mmol/l母液。
[0041]
步骤5、将实验分为模型组(不加药物,加酶、磷酸钾缓冲液和底物)、实验组(新化合物)、对照组、空白组和药物组,其中对照组为iso
‑
1,不同组的药物均设置有5个浓度梯度(100μmol/l、30μmol/l、10μmol/l、3μmol/l、1 μmol/l),以上每组均设有3个复孔,见表2。
[0042]
表2:模型组、实验组、对照组、空白组及药物组添加的试剂
[0043][0044][0045]
步骤6、按照表2所列添加试剂从上到下的顺序及用量进行各组实验,实验组及对照组实验吸光度测定步骤为:将10μl不同浓度的药物、10μlmif酶溶液 (120nm)、50μl10mmol/l磷酸钾缓冲液(0.5mm edta,ph=6.2),混合,摇匀, 25℃预温育30min,加入30μl l
‑
多巴色素甲酯后,去除孔中气泡,立即用酶标仪在475nm处测定其吸光度值;并按前述步
骤进行模型组、空白组(不加酶和药物,加磷酸钾缓冲液和底物)和药物组(不加酶,加药物新化合物或iso
‑
1、磷酸钾缓冲液和底物)实验的吸光度测定。
[0046]
表3新化合物对mif酶的抑制活性
[0047]
化合物编号ic50(μm)新化合物12.7iso
‑
115.2
[0048]
其中,ic50(μm)为酶活性抑制率为50%时新化合物的浓度,用于表示对mif 酶的抑制活性;iso
‑
1为mif酶的阳性对照药。
[0049]
如表3所示,新化合物对mif酶具有较好的抑制活性。
[0050]
为更好理解新化合物与mif的结合模式,现采用分子对接方法进行验证和阐释:
[0051]
步骤1、数据库有uniprot数据库(https://www.uniprot.org/)、pdb数据库 (http://www.rcsb.org/);软件有chemoffice2010(美国perkinelmer公司)、 sybyl 1.0软件(美国tripos公司)、discovery studio 2017 r2 client(ds,美国accelrys公司开发)、pymol。
[0052]
步骤2、运用chemdraw软件画出新化合物的结构式,保存为mol2格式。将新化合物的mol2格式结构式导入sybyl 1.0软件,采用分子力学程序 minimize进行结构优化,赋予tripos力场及加载gasteiger
‑
huckel电荷,优化后得到的稳定构象保存为mol2格式,建立配体小分子化合物库,为分子对接做准备。
[0053]
步骤3、从pdb数据库(http://www.rcsb.org)下载目标靶蛋白巨噬细胞移动抑制因子(pdb id:3l5s)的晶体结构,对靶蛋白用application中的docking 模块进行修饰、加氢及加载amber ff99电荷,并根据靶蛋白复合物中配体确定对接的活性位点,将处理后的蛋白保存,为后续分子对接研究做准备。
[0054]
步骤4、利用sybyl 1.0软件surflex
‑
dock模块将配体小分子化合物库和靶蛋白进行分子对接,对接的结果以打分函数total score给出,以mol2格式保存。利用sybyl分子对接模块的total score打分函数对配体分子进行筛选,totalscore打分函数综合考虑了极性作用、疏水作用、焓和溶剂化等因素,该值越大,对接复合物越稳定,说明小分子化合物与大分子蛋白质的匹配结合作用越好。
[0055]
步骤5、采用discovery studio软件中receptor
‑
ligand interactions模块对分子对接结果进行分析,并制作三维和二维效果图。
[0056]
表4新化合物与靶蛋白的对接得分
[0057][0058]
新化合物与mif(pdb id:3l5s)的对接得分如表4所示。
[0059]
mif是一个同源三聚体,并且与已知的任何细胞因子没有结构同源性。每个单体有115个氨基酸组成,包含2个反向平行的α螺旋和6个β折叠,6个折叠中的4个构成一个β片层,3个单体围绕成圆筒型的溶剂可及通道。通过对新化合物与mif的结合模式进行研究(图10),新化合物与氨基酸残基lys
‑
32、tyr
‑
36、 ile
‑
64、trp
‑
108、gln
‑
35、tyr
‑
95和ser
‑
63形成了氢键相互作用,同时与phe
‑
113、 trp
‑
108、tyr
‑
36、phe
‑
49之间具有疏水作用力,与mif
形成了紧密的结合。这些氢键和疏水相互作用在新化合物与mif的结合中发挥了重要作用。因此,结合新化合物与mif的对接得分,说明本发明的新化合物与靶蛋白具有较好的结合活性,新化合物可能是mif酶的潜在抑制剂。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。