一种2,5
‑
噻吩二羧酸的制备工艺
技术领域
1.本发明涉及药物合成技术领域,特指一种2,5
‑
噻吩二羧酸的制备工艺。
背景技术:
2.2,5
‑
噻吩二羧酸是一种重要的原料药中间体,在诸多新药合成中常有出现;现有的合成技术中,2,5
‑
噻吩二羧酸主要由原料2,5
‑
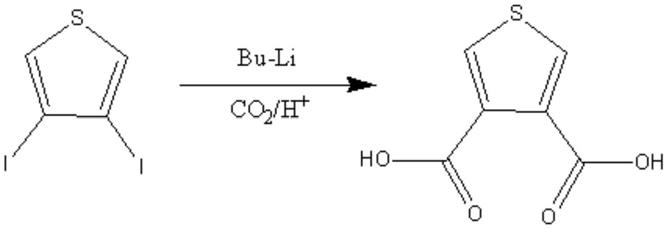
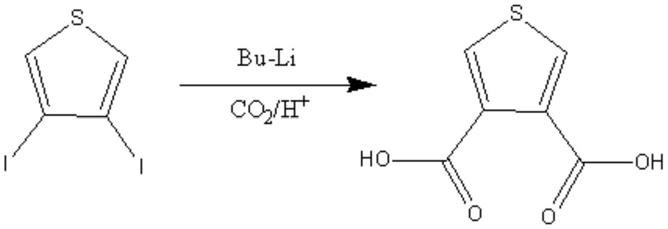
二碘噻吩与丁基锂发生锂化反应,再通入酸化的二氧化碳气体进行烷基化反应,得到产物。合成路线如下:
[0003][0004]
此方法得到的产物,不仅纯度低于60%,且涉及到的丁基锂性质很活泼,非常不稳定,大大增加了生产的危险性,具有一定的局限性。
技术实现要素:
[0005]
本发明目的是为了克服现有技术的不足而提供一种2,5
‑
噻吩二羧酸的制备工艺。
[0006]
为达到上述目的,本发明采用的技术方案是:一种2,5
‑
噻吩二羧酸的制备工艺,包含以下步骤:
[0007]
s1:将原料氰乙酸乙酯、丙酮酸甲酯、硫粉、有机溶剂加入反应瓶中,搅拌均匀,并滴加碱溶液,保持高温,搅拌定量时间;
[0008]
s2:将步骤1中获得的反应液冷却至室温,加入淬灭剂淬灭,再加入饱和食盐水,搅拌至出现大量沉淀后过滤,得到的滤渣以溶剂清洗,并烘干,得到中间产物2
‑
氨基噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯;
[0009]
s3:在反应装置中加入氧化剂和有机溶剂,搅拌均匀,将体系升至40℃,并缓慢滴加步骤2中获得的中间产物2
‑
氨基噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯的有机溶剂溶液,控制滴速,保持60℃,搅拌至tlc显示反应结束;
[0010]
s4:将步骤3中获得的反应液降低至室温,减压浓缩去除溶剂后,减压蒸馏,收集固定温度的馏分,得到中间产物噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯;
[0011]
s5:将步骤4中获得的中间产物噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯、碱溶液加入反应瓶中,搅拌均匀,加热至80℃,搅拌至tlc显示反应结束,降低体系至室温,并用酸调节体系的酸碱度,有大量不溶物析出,过滤后,得到最终产品2,5
‑
噻吩二羧酸。
[0012]
优选的,步骤s1中,所述氰乙酸乙酯与硫粉的摩尔比为1:0.5~1:3。
[0013]
优选的,步骤s1中,所述碱为氢氧化钠、碳酸钾、三乙胺中的一种或者几种。
[0014]
优选的,步骤s1中,所述高温为40~100℃,搅拌时间为10~24h。
[0015]
优选的,步骤s2中,所述淬灭剂为乙醇、水、冰水中的一种或者几种。
[0016]
优选的,步骤s2中,所述溶剂为乙醇、乙酸乙酯、二氯甲烷、水中的一种或者几种。
[0017]
优选的,步骤s3中,所述反应装置为冷凝管、鼓泡器、索氏提取器中的一种或者几种。
[0018]
优选的,步骤s3中,所述氧化剂为亚硝酸叔丁酯、高锰酸钾、双氧水中的一种或者几种。
[0019]
优选的,步骤s1和s3中,所述有机溶剂为乙酸乙酯、二氯甲烷、四氢呋喃、甲醇、dmf中的一种或者几种。
[0020]
优选的,步骤s4中,收集的馏分为90~110℃、110~150℃、150~200℃中的一种或者几种。
[0021]
优选的,步骤s5中,所述碱溶液为2n的氢氧化钠溶液、1n的三乙胺溶液、10n的氢氧化钾溶液中的一种或者几种。
[0022]
优选的,步骤s5中,所用酸为1n盐酸、5n硫酸、10n三氟醋酸中的一种或者几种。
[0023]
优选的,步骤s5中,调节体系酸碱至ph为1~2、2~3、5~6中的一种。
[0024]
由于上述技术方案的运用,本发明与现有技术相比具有下列优点:
[0025]
本发明所涉及原料价格低廉,反应温和且收率比较高,不需要过硅胶柱提纯,非常适合放大生产。
附图说明
[0026]
下面结合附图对本发明技术方案作进一步说明:
[0027]
附图1为本发明实施例3中所述的2,5
‑
噻吩二羧酸的制备工艺的核磁图。
具体实施方式
[0028]
下面结合附图及具体实施例对本发明作进一步的详细说明。
[0029]
本发明的合成路线如下:
[0030][0031]
实施例1:
[0032]
将原料氰乙酸乙酯(200g,1.77mol)、丙酮酸甲酯(200g,1.96mol)、硫粉(39.65g,
1.24mol)、dmf(1l)依次加入反应瓶中,搅拌均匀,保持搅拌并缓慢加入氢氧化钠固体(106.20g,2.66mol),保持体系为50℃,搅拌反应10h;
[0033]
将反应液冷却至室温,先加入无水乙醇(5l)淬灭,再加入3l饱和食盐水,反应瓶中出现大量的沉淀,然后过滤,以蒸馏水(500ml)清洗滤渣,重复3次,并烘干,得到中间产物2
‑
氨基噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯;
[0034]
在添加了冷凝管的三颈烧瓶中分别加入亚硝酸叔丁酯(87.71g,0.85mol)和乙酸乙酯(500ml),搅拌均匀,并将体系升至40℃,然后缓慢滴加中间产物2
‑
氨基噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯(150g,0.65mol)的乙酸乙酯(200ml)溶液,控制滴速,保持60℃,搅拌至tlc显示反应结束;
[0035]
反应液降低至室温,减压浓缩去除溶剂后,减压蒸馏,收集90~110℃的馏分,得到中间产物噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯。
[0036]
将中间产物噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯(150g,0.70mol)、2n的氢氧化钠溶液(1.05l,2.10mol)加入反应瓶中,搅拌均匀,并加热至80℃,搅拌至tlc显示反应结束,降低体系至室温,最后用1n的稀盐酸调节体系的酸碱度至1~2,过滤后,得到产品2,5
‑
噻吩二羧酸。
[0037]
此实施例1得到的纯度只有80%左右,收率76%。
[0038]
实施例2:
[0039]
将原料氰乙酸乙酯(200g,1.77mol)、丙酮酸甲酯(200g,1.96mol)、硫粉(169.92g,5.314mol)、甲醇(1l)依次加入反应瓶中,搅拌均匀,保持搅拌并缓慢加入三乙胺(269.17g,2.66mol),保持体系为100℃,搅拌反应24h;
[0040]
将反应液冷却至室温,先加入冰水(5l)淬灭,再加入3l饱和食盐水,反应瓶中出现大量的沉淀,然后过滤,以蒸馏水(500ml)清洗滤渣,重复3次,并烘干,得到中间产物2
‑
氨基噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯;
[0041]
在添加了索氏提取器的三颈烧瓶中分别加入30%的双氧水(80ml,0.85mol)和四氢呋喃(500ml),搅拌均匀,并将体系升至40℃,然后缓慢滴加滴加中间产物2
‑
氨基噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯(150g,0.65mol)的四氢呋喃(200ml)溶液,控制滴速,保持60℃,搅拌至tlc显示反应结束;
[0042]
反应液降低至室温,减压浓缩去除溶剂后,减压蒸馏,收集150~200℃的馏分,得到中间产物噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯;
[0043]
将中间产物噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯(150g,0.70mol)、10n的氢氧化钾溶液(0.21l,2.10mol)加入反应瓶中,搅拌均匀,并加热至80℃,搅拌至tlc显示反应结束,降低体系至室温,最后用10n的三氟乙酸调节体系的酸碱度至5~6,过滤后,得到产品2,5
‑
噻吩二羧酸。
[0044]
此实施例2得到的产品纯度只有85%左右,收率70%。
[0045]
实施例3:
[0046]
将原料氰乙酸乙酯(200g,1.77mol)、丙酮酸甲酯(200g,1.96mol)、硫粉(56.64g,1.77mol)、二氯甲烷(1l)依次加入反应瓶中,搅拌均匀,保持搅拌并缓慢加入碳酸钾固体(367.61g,2.66mol),保持体系为80℃,搅拌反应18h;
[0047]
将反应液冷却至室温,先加入水(5l)淬灭,再加入3l饱和食盐水,反应瓶中出现大
量的沉淀,然后过滤,以蒸馏水(500ml)清洗滤渣,重复3次,并烘干,得到中间产物2
‑
氨基噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯;
[0048]
在添加了鼓泡器和冷凝管的三颈烧瓶中分别加入高锰酸钾固体(134.33g,0.85mol)和二氯甲烷(500ml),搅拌均匀,并将体系升至40℃,然后缓慢滴加滴加中间产物2
‑
氨基噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯(150g,0.65mol)的二氯甲烷(200ml)溶液,控制滴速,保持60℃,搅拌至tlc显示反应结束;
[0049]
反应液降低至室温,减压浓缩去除溶剂后,减压蒸馏,收集110~150℃的馏分,得到中间产物噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯;
[0050]
将中间产物噻吩
‑
3,4
‑
二羧酸3
‑
乙酯4
‑
甲酯(150g,0.70mol)、三乙胺(212.50g,2.10mol)加入反应瓶中,搅拌均匀,并加热至80℃,搅拌至tlc显示反应结束,降低体系至室温,最后用5n的硫酸调节体系的酸碱度至2~3,过滤后,得到产品2,5
‑
噻吩二羧酸。
[0051]
此实施例3得到的产品纯度95%左右,收率90%,如图1所示。
[0052]
本发明所涉及原料价格低廉,反应温和且收率比较高,不需要过硅胶柱提纯,非常适合放大生产。
[0053]
以上仅是本发明的具体应用范例,对本发明的保护范围不构成任何限制。凡采用等同变换或者等效替换而形成的技术方案,均落在本发明权利保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。