1.本发明属于不对称催化合成领域,涉及一种轴手性双膦配体及其制成的催化剂,以及催化剂在不对称氢化反应和类似反应中的应用。
背景技术:
2.随着人们对光学纯的手性药物、农药、香料和其他精细化学品的需求日益增加,不对称催化技术获得了快速的发展。由于氢化反应具有高效、高选择性、高原子经济性的优点,不对称氢化反应一直是不对称催化技术领域的研究热点。对前手性的烯烃、酮和亚胺等不饱和化合物进行不对称氢化还原,是获得手性化合物片段的最为高效的方法之一。影响不对称氢化效率的核心因素是过渡金属和手性配体的有效组合,但由于元素周期表的限制,可供选择的过渡金属十分有限,因此新型手性配体的设计和研发已经成为不对称氢化领域中最核心的研究主题,贯穿整个不对称氢化的发展历史。
3.1965年wilkinson教授报道了第一例均相催化的氢化反应,随后在1968年knowles教授报道了第一例均相催化的不对称氢化反应。由于具有高效、高选择性、高原子经济性的优点,不对称催化氢化反应在所有已经实现工业化生产的不对称催化反应中占据70%以上。另一方面,在不对称氢化反应中,手性配体的结构对反应的活性和立体选择性具有重要的影响,因此化学家们可以通过合理的配体电性和空间位阻设计实现对反应的精细调控。然而并没有一种配体能够解决所有的问题,因而发展高效、高选择性、底物适用范围广的手性配体和不对称催化体系将是这一研究领域的永恒主题。
4.自knowles教授的dipamp配体和noyori教授的binap配体报道以来,各种骨架的双膦配体受到了广泛地关注和研究,张绪穆教授在这方面做出了杰出的贡献,发展了一系列高效的膦手性和骨架手性的双齿膦配体,建立了一个非常有用不对称氢化手性配体工具箱。但是目前能应用于工业化生产的手性膦配体屈指可数,而且大多手性双膦配体的合成路线复杂,因此设计合成新的手性膦配体,具有十分重要的意义。几种代表性的双齿膦配体骨架如下:
[0005][0006]
相较于已有的轴手性双膦配体,本发明提出了一个原料易得、合成路线简单、收率高、易于大规模制备、结构和电性便于调节的新型轴手性双膦配体,该配体已在催化不对称
氢化反应中表现出超高的反应活性和立体选择性,具有广阔的工业应用前景。
技术实现要素:
[0007]
本发明的目的是提供一种结构新颖的轴手性双膦配体,另一方面提供由该配体制备而得的催化剂及其在催化不对称氢化反应及其类似反应上的用途。该双膦配体在空气气氛中稳定,催化活性高,立体选择性高,易于实现工业化生产。为了实现上述目标,本发明提出的轴手性双膦配体具有以下通式(i)的结构:
[0008][0009]
通式(i)中:
[0010]
r1、r2独立为卤素所取代或者未取代的烷基、烷氧基、芳基或者氢原子;r3、r4独立为烷基、烷氧基、芳基、芳氧基或者氢原子,r3和ra成环或者不成环。
[0011]
该配体的合成用到的起始原料是便宜易得的(1r,2r)-1,2-环己烷二甲酸,仅通过四步反应即可得到目标配体。合成路线中避免了困难的拆分步骤,具有重要的潜在应用价值。配体合成路线短,原料便宜易得,说明此类配体具有良好的工业应用前景。
[0012]
作为本发明的一种优选技术方案,r1、r2独立为卤素所取代或者未取代的c
1-6
烷基、c
1-6
烷氧基、苯基、或者氢原子;r3、r4独立为卤素所取代或者未取代的c
1-6
烷基、c
3-7
环烷基、c
1-6
烷氧基、苯基、或者氢原子。
[0013]
所述卤素选自氟、氯、溴、碘;
[0014]
所述c
1-6
的烷基选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、仲戊基、叔戊基、正己基、异己基、新己基、仲己基、叔己基;
[0015]
所述c
1-6
的烷氧基选自甲氧基、乙氧基、丙氧基、异丙氧基、正丁氧基、异丁氧基、仲丁氧基、叔丁氧基、正戊氧基、异戊氧基、新戊氧基、仲戊氧基、叔戊氧基、正己氧基、异己氧基、新己氧基、仲己氧基、叔己氧基;
[0016]
所述c
3-7
环烷基选自环丙基、环丁基、环戊基、环己基、环庚基。
[0017]
以下给出了本发明轴手性配体部分实例(l1-l15),每一个配体对应两种对映异构体,也是本发明的内容,具体如下:
[0018][0019][0020]
本发明还提供所述轴手性双膦配体的合成方法:
[0021][0022]
本发明还提供催化不对称反应的催化剂,由上述本发明提供的新型轴手性双膦配体与过渡金属前体络合而成。
[0023]
本发明还提供了催化剂的制备方法:本发明的轴手性双膦配体与过渡金属前体在合适的溶剂中络合反应若干时间。同时还提供了该催化剂在简单酮的不对称氢化反应中的应用案例。
[0024]
合适的过渡金属有ru、rh、ir、fe、co、ni、mn、cu、ag或pd。
[0025]
合适的过渡金属前体包括[rh(nbd)2]
bf
4-;[rh(nbd)cl]2;[rh(cod)cl]2;[rh(cod)2]
x-;rh(acac)(co)2;rh(ethylene)2(acac);[rh(ethylene)2cl]2;rhcl(pph3)3;rh(co)2cl2;ru(arylgroup)x2;ruhx(l)2(diphosphine);rux2(l)2(diphosphine);ru(arene)x2(diphosphine);ru(rcoo)2(diphosphine);ru(methallyl)2(diphosphine);ru(aryl group)x2(pph3)3;ru(cod)(cot);ru(cod)(cot)x;rux2(cymene);ru(cod)n;rucl2(=chr)(pr'3)2;ru(arylgroup)x2(diphosphine);rucl2(cod);[ru(cod)2]x;rux2(diphosphine);ru(arh)cl2;ru(cod)(methallyl)2;ir(nbd)cl]2;[ir(nbd)2]x;[ir(cod)cl]2;[ir(cod)2]x;[ni(allyl)x]2;ni(acac)2;ni(cod)2;nix2;mnx2;mn(acac)2;cox2;fex2;cux;cux2;agx;[pd(allyl)cl]2;pdcl2;pd(oac)2;pd(cf3coo)2。
[0026]
以上过渡金属前体中,r和r'可分别为烷基、烷氧基或取代烷基,aryl为芳基,x为负阴离子,如cl-、br-、bf
4-、clo
4-、sbf
6-、pf
6-、cf3so
3-、rcoo-、b(ar)
4-,其中ar可为3,5-二三氟甲基苯或氟苯。l是溶剂分子,如ch3cn等。
[0027]
作为本发明的一种优选技术方案,所述催化剂选自:
[0028][0029]
本发明中涉及到的新型配体和过渡金属络合而成的催化剂,可用于多种不同类型的不对称催化反应,如:不对称氢化反应、不对称转移氢化反应、不对称硅氢化反应、不对称硼氢化反应、不对称氢甲酰化反应、烯丙基化反应、烯烃复分解反应、异构化反应、diels-alder反应、不对称偶联反应、aldol反应、michael加成反应、不对称环氧化反应、动力学拆分和[m n]环化反应等。
[0030]
例如,所述催化剂应用于催化芳香酮、脂肪酮、环状α-脱氢胺基酮的反应中,具有良好的催化效率和收率。
[0031]
本发明相对于现有技术的有益效果包括:
[0032]
本发明提供的轴手性双膦配体,相对于binap等双膦配体,无论在空间位阻还是电性上都非常易于调控,可能具有更广泛地底物适用范围。并且该配体的合成可从廉价易得的(1r,2r)-1,2-环己烷二甲酸经过简单的四步反应快速获得,具有空气中十分稳定,易于纯化的特点,非常适于大规模合成制备,有很好的工业化应用前景。
具体实施方式
[0033]
下面结合具体实施例对本发明做进一步的说明,但本发明不仅限于以下实施例。
[0034]
实施例1:
[0035]
配体l1的合成:
[0036][0037]
在0℃下,将3.04g氢化铝锂(80mmol)分批溶于40ml无水thf中,分批加入3.44g(1r,2r)-1,2-环己烷二甲酸(20mmol),升温至80℃回流反应16小时。反应结束后,加入5ml水淬灭反应,加入20ml,过滤,收集有机相,无水硫酸钠干燥,减压除去溶剂,得到无色油状液体2.3g,收率为81%。
[0038][0039]
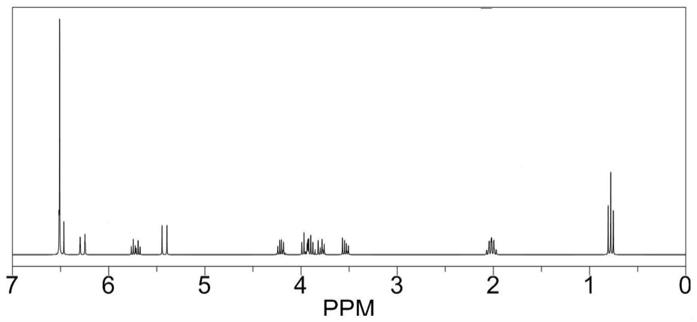
称取28.8g(1r,2r)-1,2-环己烷二甲醇(0.2mol)起始原料,用30ml二氯甲烷溶解,再加入84ml三乙胺(0.6mol),在-5℃搅拌下滴加34ml甲基磺酰氯(50.4g,0.44mol),缓慢升至室温,继续反应2个小时。用薄层色谱监测(tlc),待反应结束后,旋蒸除去溶剂,依次用2m稀盐酸,饱和碳酸氢钠溶液和饱和食盐水洗涤,再经过无水硫酸钠干燥,减压除去溶剂,得到黄色油状液体。继续向反应瓶中加入120g碘化钠(0.8mol)和500ml丙酮,在70℃下回流12个小时。待反应结束后,先加水洗净碘化钠,再用乙酸乙酯萃取,最后用干燥剂无水硫酸钠进行干燥,旋蒸除去所用到的有机溶剂,用硅胶柱色谱提纯分离,即可得粗产品。在室温下进行重结晶操作,有大量白色棒状晶体析出,收率为90%。
[0040]1h nmr(600mhz,cdcl3)δ3.31(dd,j=8.47,8.64hz,2h),3.26(s,1h),3.24(s,1h),1.78
–
1.71(m,2h),1.66
–
1.65(m,2h),1.40
–
1.31(m,4h),0.91(s,2h).
13
c nmr(151mhz,cdcl3)δ41.2,32.4,25.6,15.7.
[0041]
[0042]
称取0.56g苯乙炔(5mmol)于10ml schlenk瓶中,加入5ml超干四氢呋喃。将反应管置于-78℃低温冷浴中,滴加2.1ml正丁基锂(2.4m),继续反应4小时,做成炔基锂试剂。将0.46g化合物3(1.25mmol)溶于2.5ml四氢呋喃中,滴加炔基锂试剂,室温反应12个小时后,用氯化铵水溶液淬灭,乙酸乙酯萃取,收集有机相,减压除去溶剂,经硅胶柱纯化得到0.27g黄色油状液体,收率为70%。
[0043]1h nmr(500mhz,cdcl3)δ7.50-7.41(m,4h),7.38-7.27(m,6h),2.64-2.52(m,4h),1.98-1.91(m,2h),1.87-1.76(m,2h),1.68-1.58(m,2h),1.46-1.29(m,4h).
13
c nmr(126mhz,cdcl3)δ131.6,128.2,127.5,124.1,88.5,82.1,40.0,32.1,26.2,24.0.
[0044][0045]
称取1.17g二氯二茂锆粉末(4mmol)和1.25g化合物4(4mmol)于50ml schlenk瓶中,然后加入15ml超干四氢呋喃,将反应瓶置于-78℃冷浴中,缓慢滴加3.4ml正丁基锂(2.4m)溶液,在此温度下反应30min后,再置于室温反应2.5个小时。随后将反应液置于0℃冷浴中,将schlenk支口抽换氩气,边通氩气边倒入0.8g氯化亚铜(8mmol)粉末,加完固体后反应液呈深色,继续在0℃反应1小时,再缓慢滴加1.6ml氯化二苯基膦(9mmol),自然升温搅拌12h,tlc监测反应。反应结束后减压除去所用的有机溶剂,加水洗涤,用二氯甲烷萃取有机相,经硅胶柱色谱纯化后得到淡黄色固体粉末。向固体粉末中加入50ml乙二胺常温搅拌5小时,再加水洗,二氯甲烷萃取分液,有机相用无水硫酸钠干燥,减压除去溶剂,用硅胶柱色谱提纯分离拿到白色固体粉末1.64g,收率为60%。
[0046]1h nmr(400mhz,cdcl3)δ7.69(dtq,j=6.7,3.4,1.9hz,4h),7.48
–
7.41(m,4h),7.41
–
7.33(m,2h),7.21(dtd,j=8.2,3.3,1.3hz,4h),7.13
–
7.05(m,2h),7.05
–
6.97(m,6h),6.96(dd,j=4.0,2.4hz,4h),6.84
–
6.75(m,4h),2.43-2.41(dd,j=12.7,2.9hz,2h),1.82
–
1.63(m,2h),1.68
–
1.66(m,2h),1.49
–
1.46(m,2h),1.29
–
1.14(m,4h),1.02
–
0.84(m,2h).
13
c nmr(101mhz,cdcl3)δ154.4
–
154.0(m),140.0(t,j=2.1hz),138.9
–
138.6(m),138.1
–
137.5(m),134.0(t,j=11.4hz),133.3(t,j=9.8hz),132.2
–
131.8(m),128.2(t,j=2.7hz),127.8,127.6
–
126.7(m),125.6,46.4,43.5,33.7,26.0.
31
p nmr(162mhz,cdcl3)δ-4.32.hrms(esi ),m/z 683.2984([m h]
),calcd for[m h]
:682.2918.[α]
23d
=-245.6(c=0.5,chcl3).
[0047]
实例2:
[0048]
配体l2的合成:
[0049][0050]
称取1.17g二氯二茂锆粉末(4mmol)和1.47g化合物5a(4mmol)于50ml schlenk瓶中,然后加入15ml超干四氢呋喃,将反应瓶置于-78℃冷浴中,缓慢滴加3.4ml正丁基锂(2.4m)溶液,在此温度下反应30min后,再置于室温反应2.5个小时。随后将反应液置于0℃冷浴中,将schlenk支口抽换氩气,边通氩气边倒入0.8g氯化亚铜(8mmol)粉末,加完固体后反应液呈深色,继续在0℃反应1小时,再缓慢滴加1.6ml氯化二苯基膦(9mmol),自然升温搅拌12h,tlc监测反应。反应结束后减压除去所用的有机溶剂,加水洗涤,用二氯甲烷萃取有机相,经硅胶柱色谱纯化后得到淡黄色固体粉末。向固体粉末中加入50ml乙二胺常温搅拌5小时,再加水洗,二氯甲烷萃取分液,有机相用无水硫酸钠干燥,减压除去溶剂,用硅胶柱色谱提纯分离拿到白色固体粉末1.98g,收率为67%。
[0051]1h nmr(400mhz,cdcl3)δ7.68-7.64(m,4h),7.41-7.36(dt,m,6h),7.16-7.12(m,4h),7.07(dd,j=9.2,5.1hz,2h),7.05
–
6.93(m,4h),6.58(s,2h),6.32(s,4h),2.50-2.32(m,2h),2.04(s,12h),1.67(h,j=9.2,7.9hz,4h),1.53-1.44(m,2h),1.24(d,j=10.1hz,4h),0.91(q,j=8.8,8.0hz,2h).
13
c nmr(101mhz,cdcl3)δ153.6-153.2(m),139.4,139.3-138.9(m),138.4-138.0(m),136.2,134.2-133.5(m),132.3-131.9(m),128.1(t,j=2.9hz),128.0-126.8(m),46.3,43.4,33.6,26.1,21.1.
31
p nmr(162mhz,cdcl3)δ-4.05.hrms(esi ),m/z 739.3611([m h]
),calcd for[m h]
:738.3544.[α]
23d
=-130.8(c=0.5,chcl3)
[0052]
实例3
[0053]
配体l3的合成:
[0054][0055]
称取1.17g二氯二茂锆粉末(4mmol)和0.75g化合物5b(4mmol)于50ml schlenk瓶中,然后加入15ml超干四氢呋喃,将反应瓶置于-78℃冷浴中,缓慢滴加3.4ml正丁基锂(2.4m)溶液,在此温度下反应30min后,再置于室温反应2.5个小时。随后将反应液置于0℃冷浴中,将schlenk支口抽换氩气,边通氩气边倒入0.8g氯化亚铜(8mmol)粉末,加完固体后反应液呈深色,继续在0℃反应1小时,再缓慢滴加1.6ml氯化二苯基膦(9mmol),自然升温搅拌12h,tlc监测反应。反应结束后减压除去所用的有机溶剂,加水洗涤,用二氯甲烷萃取有机相,经硅胶柱色谱纯化后得到淡黄色固体粉末。向固体粉末中加入50ml乙二胺常温搅拌5
小时,再加水洗,二氯甲烷萃取分液,有机相用无水硫酸钠干燥,减压除去溶剂,用硅胶柱色谱提纯分离拿到白色固体粉末0.89g,收率为40%。
[0056]1h nmr(400mhz,cdcl3)δ7.33(t,j=7.0hz,4h),7.2-7.25(m,8h),6.96-7.10(m,8h),2.43-2.41(m,2h),2.20(s,6h),1.82
–
1.63(m,2h),1.68
–
1.66(m,2h),1.49
–
1.46(m,2h),1.29
–
1.14(m,4h),1.02
–
0.84(m,2h),1.67(m,4h),1.53-1.44(m,2h),1.24(d,j=10.1hz,4h),0.91(q,j=8.8,8.0hz,2h)..
31
p nmr(162mhz,cdcl3)δ-4.2.
[0057]
实施例4:
[0058]
配体l1和金属ru前体络合合成催化剂i
[0059][0060]
称取407mg[rucl2(benzene)]2(0.814mmol)和1.11g配体l1(1.63mmol)置于20ml schlenk瓶中,置换氩气,然后加入12ml提前鼓泡除氧过的dmf。将反应管置于100℃油浴中加热1小时,反应液呈暗红色。将反应体系冷却至25℃,继续加入346mg(s,s)-dpen(1.63mmol),在室温下继续搅拌反应6小时。减压除去溶剂,反应残渣用正己烷洗涤,干燥得到亮黄色固体粉末1.2g,收率为70%。
[0061]
实施例5:
[0062]
配体l3和金属ru前体络合合成催化剂ii
[0063][0064]
称取407mg[rucl2(benzene)]2(0.814mmol)和0.91g配体l3(1.63mmol)置于20ml schlenk瓶中,置换氩气,然后加入12ml提前鼓泡除氧过的dmf。将反应管置于100℃油浴中加热1小时,反应液呈暗红色。将反应体系冷却至25℃,继续加入346mg(s,s)-dpen(1.63mmol),在室温下继续搅拌反应6小时。减压除去溶剂,反应残渣用正己烷洗涤,干燥得到亮黄色固体粉末1.0g,收率为67%。
[0065]
实施例6:
[0066]
催化剂效果考察及条件筛选
[0067][0068]
在氩气氛围的手套箱内,向5ml氢化小瓶中依次加入0.23ml底物苯乙酮6(2mmol),80ul碱的异丙醇溶液(0.25mmol/l,0.02mmol),0.2mol%催化剂(0.004mmol),最后加入2ml异丙醇溶剂。将氢化小瓶放入氢化反应釜中,氢化置换反应釜内气体三次后充入10atm氢
气,在室温下反应12小时。反应完毕释放氢气后,反应混合液用硅胶短柱纯化,浓缩滤液真空干燥得到油状液体,即苯乙醇7。hplc测得转化率和ee值,结果如下表1所示。
[0069]
表1.
[0070][0071]
实施例7:
[0072]
配体l2和[rh(nbd)cl]2原位络合催化环状α-脱氢胺基酮的不对称催化氢化反应
[0073][0074]
在氩气氛围下,称取金属前体[rh(nbd)cl]2(0.46mg,0.001mmol)和配体l2(0.81mg,0.0011mmol)置于样品瓶中,再加入0.5ml甲醇溶液,常温搅拌2小时络合。在5ml氢化瓶里称取0.2mmol化合物8a-p,往氢化瓶中依次加入配好的催化剂(1mol%)和0.5ml甲醇溶液,再搅拌10min直至固体完全溶解,然后将氢化瓶小心放入高压反应釜中,先用氢气把反应釜中的气体置换三次,然后充入10atm的氢气,在25℃下反应24个小时。反应结束后,小心释放高压反应釜中的氢气,减压除去溶剂,经硅胶柱色谱纯化后,得到纯的氢化产物9a-p,反应收率和对映选择性ee值的结果如下:
[0075][0076]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。