cd73抑制剂的结晶形式
1.本发明涉及5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的结晶形式,和包含它们的药物组合物,其抑制cd73的活性,并且可用于治疗癌症。
[0002]
cd73,也称为5
’‑
核苷酸酶或外-5’核苷酸酶(ec 3.1.3.5),是将5’单核苷酸转化为核苷的酶。cd73在许多组织中表达,并且在癌组织中上调,并且cd73途径通过腺苷受体信号传导限制抗肿瘤t细胞免疫来促进肿瘤生长(zhang b,cancer research 2010;70:6407-6411;antonioli l等人,drug discovery today 2017;22:1686-1696)。cd73-缺陷小鼠具有提高的抗肿瘤免疫,并且对试验转移有抗性(stagg j等人,cancer research 2011;71:2892-2900)。由肿瘤cd73产生的细胞外腺苷在肿瘤微环境中积累,损害抗肿瘤t细胞免疫(zhang b;antonioli l),并且与肿瘤的免疫逃避、增殖、迁移、新血管形成、转移和肿瘤细胞的化学抗性有关(inoue y等人,oncotarget 2017;8:8738-8751)。还报道了升高的cd73表达与增加的免疫抑制有关(jin d等人,cancer res.70:2245-2255(2010))。
[0003]
因此,cd73途径发挥免疫抑制作用,并且已经提示阻断cd73途径可以用于治疗癌症(allard d等人,immunotherapy 2016;8:145-163)。cd73抑制剂用于治疗癌症的临床试验(hay cm等人,oncoimmunol.2016;5(8):e1208875;allard d)。
[0004]
名称为“cd73抑制剂”的美国申请16/481,146是国际申请号pct/us2019/019074的美国国家阶段,并且公开了某些cd73抑制剂分子,包括化合物:
[0005][0006]
(式i,其为5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮)。
[0007]
因此,本发明提供了以下化合物的结晶形式
[0008][0009]
(5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮)。
[0010]
在优选的实施方案中,结晶形式是以下化合物的结晶形式
[0011][0012]
(5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮。
[0013]
本发明还提供了药物组合物,其包含化合物5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的结晶形式,以及一种或多种药学上可接受的载体、稀释剂或
赋形剂。在优选的实施方案中,药物组合物包含5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的结晶形式,以及一种或多种药学上可接受的载体、稀释剂或赋形剂。
[0014]
结晶形式1
[0015]
在一个优选的实施方案中,本发明提供了组合物,其包含至少500gm下式化合物的无水结晶形式:
[0016][0017]
(5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮)。
[0018]
在另一个优选的实施方案中,组合物中的无水结晶形式的特征在于以下至少一种:(a)在x-射线衍射图中衍射角2-θ为4.7
°
的峰,与在11.7
°
、15.8
°
、16.0
°
、26.8
°
和27.2
°
处的一个或多个峰组合;衍射角的允许误差为0.2度;和(b)
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的峰:25.4、28.9、43.1、109.3、124.4、128.2和160.7ppm(分别为
±
0.2ppm)。
[0019]
在另一个优选的实施方案中,无水结晶形式的特征还在于
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的一个或多个峰:16.6、18.3、20.7、22.1、23.9、26.6、27.6、28.1、30.2、31.2、32.2、38.6、39.7、41.2、111.0、112.0、130.7、145.1、146.3、150.1、151.0、153.4、156.7、157.6、158.6、159.3、160.7、163.4、165.9、169.4和170.2ppm(分别为
±
0.2ppm)。
[0020]
在另一个优选的实施方案中,无水结晶形式的特征在于
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的峰:16.6、18.3、20.7、22.1、23.9、25.4、26.6、27.6、28.1、28.9、30.2、31.2、32.2、38.6、39.7、41.2、43.1、109.3、111.0、112.0、124.4、128.2、130.7、145.1、146.3、150.1、151.0、153.4、156.7、157.6、158.6、159.3、160.7、163.4、165.9、167.0、169.4和170.2ppm(分别为
±
0.2ppm)。
[0021]
在另一个优选的实施方案中,组合物中的无水结晶形式是化合物5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮。
[0022]
在另一个优选的实施方案中,组合物至少50%、60%、70%、80%、90%、95%、96%、97%、98%、99%或100%不含污染物。在另一个优选的实施方案中,组合物至少95%不含污染物。在另一个优选的实施方案中,组合物至少96%不含污染物。在另一个优选的实施方案中,组合物至少97%不含污染物。在另一个优选的实施方案中,组合物至少98%不含污染物。在另一个优选的实施方案中,组合物至少99%不含污染物。
[0023]
在另一个优选的实施方案中,组合物是颗粒组合物。在另一个优选的实施方案中,颗粒组合物是湿颗粒组合物。在另一个优选的实施方案中,颗粒组合物是干颗粒组合物。
[0024]
在另一个优选的实施方案中,本发明提供了生产容器,其包含至少1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900或1000kg的组合物。在另一个优选的实施方案中,本发明提供了包含至少1kg的组合物的生产容器。在另一个优选的实施方案中,本发明提供了包含至少5kg的组合物的生产容器。
[0025]
在另一个优选的实施方案中,本发明提供了药物组合物,其包含所述组合物和一种或多种药学上可接受的载体、稀释剂或赋形剂。
[0026]
结晶形式2
[0027]
在另一个优选的实施方案中,结晶形式是下式化合物的去溶剂化(无水)结晶形式:
[0028][0029]
(5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮),其特征在于以下中的至少一种:(a)在x-射线衍射图中衍射角2-θ为4.3-5.5
°
的峰,与在6.4
°
、9.6
°
、11.0
°
和11.6
°
处的一个或多个峰组合;衍射角的允许误差为0.2度;和(b)
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的峰:17.2、28.7、122.0、126.1、143.6和155.5ppm(分别为
±
0.2ppm)。
[0030]
在另一个优选的实施方案中,其中在x-射线衍射图中衍射角2-θ为4.3-5.5
°
的峰是位于衍射角2-θ为4.3
°
。
[0031]
在另一个优选的实施方案中,去溶剂化(无水)结晶形式的特征还在于
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的一个或多个峰:18.9、21.1、22.5、24.4、26.7、27.8、29.7、39.7、41.3、45.5、50.1、110.7、111.7、112.4、125.0、129.1、144.8、146.2、147.2、149.9、151.1、153.0、155.5、157.2、159.0、160.6、161.4、162.7、164.8、167.0、168.3和170.4ppm(分别为
±
0.2ppm)。
[0032]
在另一个优选的实施方案中,去溶剂化(无水)结晶形式的特征在于
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的峰:17.2、18.9、21.1、22.5、24.4、26.7、27.8、28.7、29.7、39.7、41.3、45.5、50.1、110.7、111.7、112.4、122.0、125.0、126.1、129.1、143.6、144.8、146.2、147.2、149.9、151.1、153.0、155.5、157.2、159.0、160.6、161.4、162.7、164.8、167.0、168.3和170.4ppm(分别为
±
0.2ppm)。
[0033]
在另一个优选的实施方案中,去溶剂化(无水)结晶形式至少50%、60%、70%、80%、90%、95%、96%、97%、98%、99%或100%不含污染物。在另一个优选的实施方案中,去溶剂化(无水)结晶形式至少95%不含污染物。在另一个优选的实施方案中,去溶剂化(无水)结晶形式至少96%不含污染物。在另一个优选的实施方案中,去溶剂化(无水)结晶形式至少97%不含污染物。在另一个优选的实施方案中,去溶剂化(无水)结晶形式至少98%不含污染物。在另一个优选的实施方案中,去溶剂化(无水)结晶形式至少99%不含污染物。
[0034]
在另一个优选的实施方案中,本发明提供了组合物,其包含至少100、200、300、400、500或1000g的去溶剂化(无水)结晶形式。在另一个优选的实施方案中,本发明提供了组合物,其包含至少100g的去溶剂化(无水)结晶形式。在另一个优选的实施方案中,组合物是颗粒组合物。在另一个优选的实施方案中,颗粒组合物是湿颗粒组合物。在另一个优选的实施方案中,颗粒组合物是干颗粒组合物。
[0035]
在另一个优选的实施方案中,本发明提供了生产容器,其包含至少1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900或1000kg
的去溶剂化(无水)结晶形式。在另一个优选的实施方案中,本发明提供了生产容器,其包含至少1kg的去溶剂化(无水)结晶形式。在另一个优选的实施方案中,本发明提供了药物组合物,其包含去溶剂化(无水)结晶形式以及一种或多种药学上可接受的载体、稀释剂或赋形剂。
[0036]
结晶形式3
[0037]
在另一个优选的实施方案中,本技术提供了下式化合物的半水合物结晶形式:
[0038][0039]
(5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮)。
[0040]
在另一个优选的实施方案中,半水合物结晶形式的特征在于以下至少一种:(a)在x-射线衍射图中衍射角2-θ为5.0
°
的峰,与在16.4
°
、17.8
°
和26.6
°
处的一个或多个峰组合;衍射角的允许误差为0.2度;和(b)
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的峰:23.5、34.6、110.2、124.1、126.8和166.2ppm(分别为
±
0.2ppm)。
[0041]
在另一个优选的实施方案中,半水合物结晶形式的特征还在于
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的一个或多个峰:16.5、18.9、19.6、20.4、22.6、24.4、26.0、27.4、28.3、30.0、30.9、38.4、40.5、110.8、130.4、131.2、146.0、147.1、150.4、151.2、153.4、157.1、157.7、158.3、159.6、163.1和169.7ppm(分别为
±
0.2ppm)。
[0042]
在另一个优选的实施方案中,半水合物结晶形式的特征在于
13
c固态nmr光谱,其包含在以下位置的参照金刚烷的高场共振(δ=29.5ppm)的峰:16.5、18.9、19.6、20.4、22.6、23.5、24.4、26.0、27.4、28.3、30.0、30.9、34.6、38.4、40.5、110.2、110.8、124.1、126.8、130.4、131.2、146.0、147.1、150.4、151.2、153.4、157.1、157.7、158.3、159.6、163.1、166.2和169.7ppm(分别为
±
0.2ppm)。
[0043]
在另一个优选的实施方案中,半水合物结晶形式至少50%、60%、70%、80%、90%、95%、96%、97%、98%、99%或100%不含污染物。在另一个优选的实施方案中,半水合物结晶形式至少95%不含污染物。在另一个优选的实施方案中,半水合物结晶形式至少96%不含污染物。在另一个优选的实施方案中,半水合物结晶形式至少97%不含污染物。在另一个优选的实施方案中,半水合物结晶形式至少98%不含污染物。在另一个优选的实施方案中,半水合物结晶形式至少99%不含污染物。
[0044]
在另一个优选的实施方案中,本发明提供了包含至少100、200、300、400、500或1000g的半水合物结晶形式的组合物。在另一个优选的实施方案中,本发明提供了包含至少100g的半水合物结晶形式的组合物。
[0045]
在另一个优选的实施方案中,组合物是颗粒组合物。在另一个优选的实施方案中,颗粒组合物是湿颗粒组合物。在另一个优选的实施方案中,颗粒组合物是干颗粒组合物。
[0046]
在另一个优选的实施方案中,本发明提供了生产容器,其包含至少1、2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900或1000kg的半水合物结晶形式。在另一个优选的实施方案中,本发明提供了生产容器,其包含至少
1kg的半水合物结晶形式。
[0047]
在另一个优选的实施方案中,本发明提供了药物组合物,其包含半水合物结晶形式以及一种或多种药学上可接受的载体、稀释剂或赋形剂。
[0048]
治疗方法
[0049]
本发明还提供了治疗癌症的方法,该方法包括给需要的患者施用有效量的化合物5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的结晶形式。
[0050]
在另一个优选的实施方案中,癌症是膀胱癌、乳癌、胆管癌、结肠直肠癌、结肠癌、胃癌、胆囊癌、胶质母细胞瘤、头颈癌、肝癌、肺癌、淋巴瘤、成神经管细胞瘤、黑素瘤、卵巢癌、胰腺癌、前列腺癌或肾癌。在一个实施方案中,乳癌是三阴性乳癌。在另一个实施方案中,肺癌是非小细胞肺癌。
[0051]
本发明还提供了化合物5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的结晶形式,其用于治疗。
[0052]
本发明还提供了化合物5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的结晶形式,其用于治疗癌症。在优选的实施方案中,结晶形式是化合物5-[5-[(1s,2s)-2-乙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮。
[0053]
本发明还提供了在制备用于治疗癌症的药物中的化合物5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的结晶形式。
[0054]
可受益于cd73抑制剂治疗的个体包括那些具有对抗pd1/pdl-1抑制剂耐药或难治的肿瘤,例如非小细胞肺癌、膀胱癌和黑素瘤;患有egfr/braf/kras突变型癌症的那些,例如非小细胞肺癌、膀胱癌、黑素瘤、结肠癌和胰腺癌;具有雌激素受体(-)癌的那些,例如三阴性乳癌;具有cd73高表达水平的个体,例如胰腺癌和结肠直肠癌。如果需要,此类个体可以选择用本文所公开的化合物或其药学上可接受的盐治疗,基于通过ihc测定所测其肿瘤中高cd73表达水平的存在;或基于通过rt-pcr测定所检测到的其肿瘤中egfr和braf突变的存在;或基于通过ihc或rt-pcr测定所检测到的其肿瘤中雌激素受体的损失;或基于通过lc-ms测定所检测到的其肿瘤或血浆中高水平腺苷和amp的存在。对于药效学评估,本文所述的基于lc-ms的离体测定可用于测量cd73抑制剂对血液中amp向腺苷转化的作用。
[0055]
在优选的实施方案中,患者是其中血清cd73活性已经测定的患者。在一个优选的实施方案中,“测定cd73活性”是指测定cd73活性是否存在。测定cd73表达水平或cd73活性的方法是本领域普通技术人员已知的,例如参见s morello等人,j.trans.med.2017;15:244。在另一个优选的实施方案中,“测定cd73活性”指通过cd73来定量amp转化为腺苷的水平,并且本文提供了基于lc-ms的测定,其有助于定量cd73活性的水平。
[0056]
在另一个优选的实施方案中,患者是组织中cd73表达已被测定的患者。在另一个实施方案中,组织是肿瘤组织。测定组织中cd73表达水平的方法是本领域普通技术人员已知的,例如使用western印迹或免疫组织化学(x-r wu等人,j.surg.oncol.2012;106:130-137)。
[0057]
在一个实施方案中,本发明提供了治疗癌症的方法,包括将有效量的本文的式i化合物或其药学上可接受的盐与一种或多种抗肿瘤剂同时、分别或顺序组合施用。抗肿瘤剂的非限制性实例包括雷莫芦单抗、耐昔妥珠单抗(necitumumab)、奥拉木单抗(olaratumab)、吉西他滨、培美曲塞、galunisertib、阿贝西利(abemaciclib)、吉非替尼、维
罗非尼、达拉菲尼、曲美替尼、顺铂、卡铂、达卡巴嗪、脂质体多柔比星、多西他赛、环磷酰胺和多柔比星、诺维本、艾日布林、紫杉醇、用于注射混悬剂的紫杉醇蛋白结合颗粒、伊沙匹隆、卡培他滨、folfox(甲酰四氢叶酸、氟尿嘧啶和奥沙利铂)、folfiri(甲酰四氢叶酸、氟尿嘧啶和伊立替康)、西妥昔单抗、egfr抑制剂、raf抑制剂、b-raf抑制剂、erk抑制剂、cdk4/6抑制剂、吲哚胺2,3-双加氧酶抑制剂、tgfβ抑制剂和tgfβ受体抑制剂。
[0058]
在另一个实施方案中,本发明提供了治疗癌症的方法,包括将有效量的本文的式i化合物或其药学上可接受的盐与一种或多种免疫肿瘤剂同时、分别或顺序组合施用。在一个优选的实施方案中,免疫肿瘤剂是抗-pd-1抗体、抗-pd-l1抗体、抗-cd137激动剂抗体或抗-ctla4抗体。免疫肿瘤剂的非限制性实例包括纳武单抗、伊匹木单抗、匹地利珠单抗、帕博利珠单抗、曲美木单抗、乌瑞芦单抗、利瑞鲁单抗(lirilumab)、阿特珠单抗、德瓦鲁单抗和抗-pd-l1抗体ly3300054(其重链和轻链序列分别在wo 2017/034916和us 2017/0058033中作为seq id nos:10和11列出)。在一个优选的实施方案中,免疫肿瘤剂是抗-pd-1抗体。在另一个优选的实施方案中,免疫肿瘤剂是抗-pd-l1抗体。在另一个优选的实施方案中,化合物是5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮,并且免疫肿瘤剂是ly3300054。
[0059]
在一个实施方案中,本发明提供了治疗非小细胞肺癌的方法,包括将有效量的本文的式i化合物或其药学上可接受的盐与另外的活性剂同时、分别或顺序组合施用。在一个优选的实施方案中,另外的活性剂是奥西替尼、西妥昔单抗或阿贝西利。在另一个优选的实施方案中,另外的活性剂是奥西替尼。在另一个优选的实施方案中,另外的活性剂是西妥昔单抗。在另一个优选的实施方案中,另外的活性剂是阿贝西利。
[0060]
在另一个优选的实施方案中,本发明提供了治疗黑素瘤的方法,包括将有效量的本文的式i化合物或其药学上可接受的盐与另外的活性剂同时、分别或顺序组合施用。在一个优选的实施方案中,另外的活性剂是braf抑制剂、抗-pd-1抗体或抗-pd-l1抗体。在另一个优选的实施方案中,另外的活性剂是braf抑制剂。在另一个优选的实施方案中,另外的活性剂是抗pd-1抗体。在另一个优选实施方案中,另外的活性剂是抗pd-l1抗体。在另一个优选实施方案中,抗pd-l1抗体为ly3300054(其重链和轻链序列分别在wo 2017/034916和us 2017/0058033中作为seq id nos:10和11列出)。
[0061]
在另一个优选的实施方案中,本发明提供了治疗结肠直肠癌的方法,包括将有效量的本文的式i化合物或其药学上可接受的盐与另外的活性剂同时、分别或顺序组合施用。在一个优选的实施方案中,另外的活性剂是阿贝西利。在另一个优选的实施方案中,化合物是5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮,并且另外的活性剂是阿贝西利。
[0062]
在另一个优选的实施方案中,本发明提供了治疗胰腺癌的方法,包括将有效量的本文的式i化合物或其药学上可接受的盐与另外的活性剂同时、分别或顺序组合施用。在一个优选的实施方案中,另外的活性剂是阿贝西利。在另一个优选的实施方案中,化合物是5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮,并且另外的活性剂是阿贝西利。
[0063]
在另一个优选的实施方案中,本发明提供了治疗三阴性乳癌的方法,包括将有效量的本文的式i化合物或其药学上可接受的盐与另外的活性剂同时、分别或顺序组合施用。
在一个优选的实施方案中,化合物是5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮。
[0064]
如上所用,以及在本发明的整个说明书中,除非另有说明,以下术语应理解为具有以下含义:
[0065]“药学上可接受的载体、稀释剂或赋形剂”是本领域通常接受的用于给哺乳动物例如人递送生物活性剂的介质。
[0066]
术语“治疗”等是指包括减缓或逆转障碍的进展。这些术语还包括减轻、改善、减弱、消除或减少障碍或病症的一种或多种症状,即使障碍或病症实际上未被消除并且即使障碍或病症的进展本身未被减缓或逆转。
[0067]“有效量”是指本发明化合物或其药学上可接受的盐或含有本发明化合物或其药学上可接受的盐的药物组合物的量,其将引起患者的生物学或医学响应或治疗临床医生对患者期望的治疗效果。在一个实施方案中,化合物或其药学上可接受的盐在体外或离体cd73酶测定中抑制amp向腺苷的转化。在另一个实施方案中,化合物或其药学上可接受的盐抑制用不同剂量的化合物治疗的动物的小鼠全血中amp向腺苷的转化。
[0068]
如本文所用,术语“患者”是指人。
[0069]
有效量可由主治诊断医生(如本领域技术人员)通过使用已知技术和通过观察在类似情况下获得的结果而容易地确定。在确定患者的有效量时,主治诊断医生考虑了许多因素,包括但不限于:患者的物种;其大小、年龄和一般健康状况;所涉及的具体疾病或障碍;疾病或障碍的程度或累及或严重性;个体患者的响应;所施用的特定化合物;施用方式;施用制剂的生物利用度特性;选择的剂量方案;伴随药物的使用;以及其它相关情况。
[0070]
本发明化合物优选配制成药物组合物,其通过任何使化合物可生物利用的途径施用,包括口服、静脉内和透皮途径。最优选地,此类组合物用于口服施用。此类药物组合物及其制备方法是本领域公知的。(参见,例如remington:the science和practice of pharmacy(d.b.troy,editor,第21版,lippincott,williams&wilkins,2006)。
[0071]
本领域技术人员将理解,本发明化合物能够形成盐。本发明化合物含有碱性杂环,因此与许多无机和有机酸中的任何一种反应形成药学上可接受的酸加成盐。此类药学上可接受的酸加成盐和制备它们的常规方法是本领域公知的。参见例如p.stahl等人,handbook of pharmaceutical salts:properties,selection and use,(vcha/wiley-vch,2008)
[0072]
如本文所用,“药学上可接受的盐”是指本发明化合物的相对无毒的无机和有机盐(s.m.berge等人,“pharmaceutical salts”,journal of pharmaceutical sciences,vol 66,no.1,january 1977)。
[0073]
如本文所用,“颗粒组合物”是指颗粒形式的组合物,其在药物生产过程中是药物组合物的前体组合物。
[0074]
如本文所用,“生产容器”是指用于生产药物容器但不是用于药物化学实验室的容器。生产容器的实例包括但不限于料斗收集器、床、干燥床、制粒机床、干燥器托盘、制粒机桶和混合碗。
[0075]
式i化合物可以根据本领域公知和认识的合成方法制备。这些反应步骤的适合反应条件是本领域公知的,并且溶剂和共试剂的适当取代是本领域技术人员已知的。同样,本领域技术人员应理解,合成中间体可根据需要或期望通过多种公知技术分离和/或纯化,并
且通常,可在随后的合成步骤中直接使用多种中间体而几乎不纯化或不纯化。此外,技术人员将理解,在一些情况下,引入部分的顺序不是关键的。制备本发明化合物所需的特定步骤顺序取决于所合成的特定化合物、起始化合物和取代部分的相对容易性,这是熟练的化学家所熟知的。除非另有说明,否则所有取代基如前所定义,并且所有试剂是本领域公知和认识的。
[0076]
如本文所用,下列术语具有所示含义:“acn”是指乙腈;“dast”是指二乙基氨基三氟化硫;“dcm”是指二氯甲烷;“dmap”是指4-二甲基氨基吡啶;“dmso”或“dmso”是指二甲亚砜;“ee”是指对映异构体过量;“es/ms”是指电喷雾质谱;“etoac”是指乙酸乙酯;“et2o”是指乙醚;“fbs”是指胎牛血清;“gc-ms”是指气相色谱-质谱;“hbss”是指hank氏平衡盐溶液;“ic
50”是指半最大抑制浓度;“lah”是指氢化锂铝;“lc-es/ms”是指液相色谱电喷雾质谱;“ms”是指质谱;“meoh”是指甲醇;“mtbe”是指甲基叔丁基醚;“nbuli”是指正丁基锂;“nm”是指纳米;“nmr”是指核磁共振;“oac”是指乙酸酯;“psi”是指磅/平方英寸;“rt”是指室温或环境温度;“scx”是指强阳离子交换;“sfc”是指超临界流体色谱;“snar”是指亲核芳族取代;“tea”是指三乙胺;“thf”是指四氢呋喃;“t
r”是指保留时间;和“w/w”是指溶液中的重量/重量比。
[0077]
式i化合物可以如下列反应方案所示合成。
[0078]
方案1
[0079][0080]
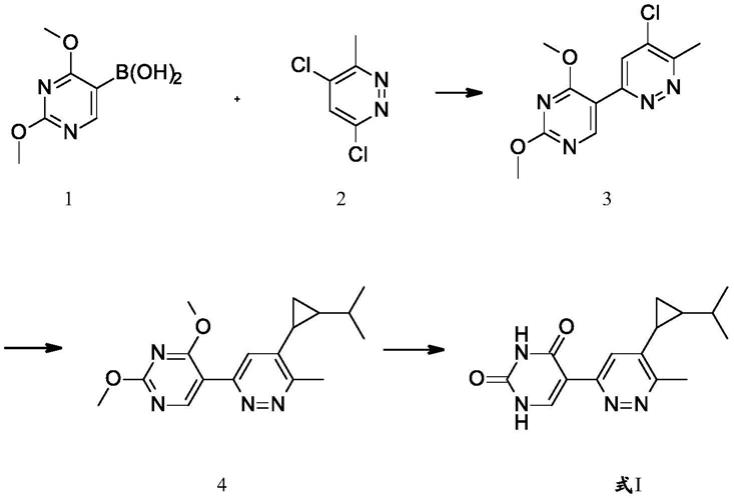
方案1描述了式i化合物的制备。使用公知的suzuki型条件,可以将市售的(2,4-二甲氧基嘧啶-5-基)硼酸1与4,6-二氯-3-甲基-哒嗪偶联。随后可以完成3的氯部分与适当取代的反式环丙基硼酸酯的suzuki偶联以获得4。技术人员将认识到化合物4的对映异构体可以使用本领域公知的手性分离技术来分离。式i化合物可以通过在本领域熟知的一系列脱甲基化条件下将4中的甲氧基脱保护而制备。
[0081]
方案2
[0082][0083]
方案2描述了制备式ii化合物所需的必要的环丙基硼酸酯。本领域技术人员将认识到,适当取代的炔5的硼氢化可以在一系列条件下进行,特别是在过渡金属催化剂例如cu或zr的存在下,以获得链烯基硼酸酯6。链烯基硼酸酯可以在本领域公知的稳定的carbenoid型条件下环丙烷化,例如simmons-smith环丙烷化,corey-chaykovsky反应,和有(例如cu、pd或ni)和没有(例如热或光化学)过渡金属催化剂的使用重氮甲烷(或重氮化合物)的环丙烷化方法,以获得适当取代的反式-环丙基硼酸酯7。本领域技术人员将认识到,热力学有利的环丙烷化产物将是7中反式对映异构体的混合物。
[0084]
方案3
[0085][0086]
方案3描述了式iii化合物的手性合成。在修饰的sandmeyer条件(基团snar)下将适当取代的氨基酸8重氮化得到9是本领域公知的。随后还原成醇10可以使用本领域公知的一系列还原剂完成,包括作为还原剂的氢化铝和二硼烷。在碱性条件下环化成手性环氧化物11在本领域中有充分描述。可通过用适合的膦酸酯(例如2-二乙氧基磷酰基乙酸乙酯或膦酰基乙酸三乙酯)和适合的碱(例如烷基锂、金属烷氧化物或金属氢化物)处理手性环氧化物11使用立体选择性horner-wadsworth-emmons反应得到反式环丙烷衍生物12(参见例如l.delhaye;a.merschaert;p.delbeke;w.brione.org.proc.res.&dev.2007,11,689-692)。水解化合物12得到相应的酸13可以在本领域充分描述的广泛的碱性条件下完成。随后,在温和的非亲核碱的存在下,使用适合的酸活化剂(例如羰基二咪唑),将酸13与适当取代的n-羟基邻苯二甲酰亚胺进行本领域中充分描述的偶联,以制备化合物14。n-羟基邻苯
二甲酰亚胺酯14的脱羧基硼化反应可以在本领域已知的许多条件下完成,包括在过渡金属催化剂(例如suzuki-miyaura反应)存在下,在光解条件下,或通过单电子转移反应,包括例如n-羟基邻苯二甲酰亚胺酯与二硼在吡啶-硼基团促进的基团偶联中的络合,以获得反式-环丙烷硼酸酯15(参见例如w.-m.cheng;s.rui;b.zhao;w.-l.xing;y.fu.org.lett.2017,19,4291-4294)。硼酸酯15与芳基氯4的偶联和随后的脱甲基化可在与方案1中所述类似的条件下进行以获得式iii的手性化合物类型。
[0087]
制备例和实施例
[0088]
下列制备例和实施例进一步说明本发明,并且代表本发明化合物的典型合成,但不应解释为以任何方式限制本发明的范围。试剂和起始材料容易获得,或者可以由本领域普通技术人员容易地合成。应当理解,制备例和实施例是通过说明而非限制的方式来阐述的,并且本领域普通技术人员可以进行多种修饰。
[0089]
lc-es/ms在agilent hp1100液相色谱系统上进行。电喷雾质谱测定(以正和/或负模式获得)在与hp1100 hplc连接的质量选择性检测器四极质谱仪上进行。lc-ms条件(低ph):柱:nx c-18 2.1
×
50mm 3.0μm;梯度:3分钟内5-100%的b,然后100%的b达0.75分钟;柱温:50℃ /-10℃;流速:1.2ml/分钟;溶剂a:含0.1%hcooh的去离子水;溶剂b:含0.1%甲酸的acn;波长214nm。可选择的lc-ms条件(高ph):柱:waters
tm
ms c-18柱2.1
×
50mm,3.5μm;梯度:5%溶剂a达0.25分钟,3分钟内梯度从5%至100%的溶剂b和100%的溶剂b达0.5分钟,或3分钟内10%至100%的溶剂b和100%溶剂b达0.75分钟;柱温:50℃ /-10℃;流速:1.2ml/分钟;溶剂a:10mm nh4hco
3 ph 9;溶剂b:acn;波长:214nm。
[0090]
在装备有质量选择性检测器质谱仪和leap自动取样器/级分收集器的agilent 1200lc-es/ms上进行制备反相色谱。在75
×
30mm上进行高ph方法,5μm粒度柱,使用10
×
20mm保护柱。流速为85ml/分钟。洗脱液是在acn中的10mm碳酸氢铵(ph 10)。
[0091]
nmr谱在bruker aviii hd 400mhz nmr光谱仪上进行,获得的cdcl3或(cd3)2so溶液以ppm表示,使用残留溶剂[cdcl3,7.26ppm;(cd3)2so,2.50ppm]作为参考标准。当报告峰多重性时,可以使用以下缩写:s(单峰)、d(双峰)、t(三重峰)、q(四重峰)、m(多重峰)、br-s(宽单峰)、dd(双双峰)、dt(双三重峰)。当报道时,偶合常数(j)以赫兹(hz)报道。
[0092]
制备例1
[0093]
4,4,5,5-四甲基-2-[(e)-3-甲基丁-1-烯基]-1,3,2-二氧杂硼杂环戊烷
[0094][0095]
历经5分钟,将频哪醇硼烷(9.5ml,64mmol)滴加到冰冷的3-甲基丁-1-炔(4.91g,72.0mmol)中。密封压力容器,温至rt并且搅拌18小时。加入双(环戊二烯基)氯化锆(iv)氢化物(2.0g,7.4mmol)和tea(1.1ml,7.9mmol)。密封压力容器,置于60℃油浴中,并且搅拌10分钟。将所得红色溶液冷却至rt达2.5小时。用dcm(200ml)稀释反应混合物,用饱和nahco3水溶液(100ml)、饱和nacl水溶液(50ml)洗涤,经mgso4干燥,通过硅胶垫(150ml)过滤,用
dcm(700ml)冲洗硅胶,并且真空浓缩,得到标题化合物(11.5g,82%)。es/ms(m/z):196(m h)。1h nmr(cdcl3)δ:1.03(d,j=6.7hz,6h),1.29(s,12h),2.37(m,1h),5.40(dd,1h),6.64(dd,1h)。
[0096]
制备例2
[0097]
外消旋-反式-2-[2-异丙基环丙基]-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷
[0098][0099]
分批将n-亚硝基-n-甲基脲加入到冰冷的et2o(70ml)和koh水溶液koh(30.5g,435mmol,70ml h2o)的两相混合物中。搅拌直至固体溶解(<5分钟)。将所得重氮甲烷溶液移液到快速搅拌的pd(oac)2(237mg,1.05mmol)和4,4,5,5-四甲基-2-[(e)-3-甲基丁-1-烯基]-1,3,2-二氧杂硼杂环戊烷(4.00g,20.4mmol)在et2o(70ml)中的冰冷混悬液中。加完后,将反应混合物温至rt,通过硅藻土过滤,并且真空浓缩滤液。将得到的残留物溶于dcm中,通过硅胶(25g)填料过滤,并且真空浓缩,得到标题化合物(4.32g,》99%)。es/ms(m/z):210(m h)。1h nmr(cdcl3)δ:-0.35(m,1h),0.45(m,1h),0.65(m,1h),0.78(m,1h),0.98(m,7h),1.23(s,12h)。
[0100]
制备例3
[0101]
4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-哒嗪
[0102][0103]
在压力瓶中,将2,4-二甲氧基-5-嘧啶基硼酸(6.75g,36.7mmol)、4,6-二氯-3-甲基-哒嗪(5.98g,36.7mmol)、[1,1
’‑
双(二苯基膦基)二茂铁]二氯钯(ii)(0.55g,0.73mmol)、cs2co3(29.9g,91.8mmol)在1,4-二烷/h2o(151ml)的4:1混合物中合并。抽空空气并且用n2回填。密封容器并且在70℃加热3小时。经硅藻土过滤残留物,并且用etoac冲洗。用水洗涤有机混合物,然后用饱和nacl水溶液洗涤,经mgso4干燥,并且蒸发至干。通过硅胶色谱纯化所得黑色残留物,使用330g柱,梯度为0-30%dcm/(在dcm中的33%meoh),历经15分钟,流速为200ml/分钟,蒸发色谱级分后,得到标题化合物(6.2g,63%)。es/ms(m/z)(
35
cl/
37
cl)267/269[m 1]
h nmr(d
6-dmso)δ:2.74(s,3h),4.00(s,3h),4.03(s,3h),8.20(s,1h),8.87(s,1h)。
[0104]
制备例3的可选择的方法
[0105]
使氮气流通过(2,4-二甲氧基嘧啶-5-基)硼酸(85g,439mmol)、4,6-二氯-3-甲基-哒嗪(75g,437mmol)和cs2co3(358g,1099mmol)在1,4-二烷(1175ml)和h2o(340ml)中的混合物5分钟。加入[1,1
’‑
双(二苯基膦基)二茂铁]二氯钯(ii)(6.6g,8.7mmol),并且将得到的混合物在75℃搅拌16小时。将反应冷却至rt,将混合物通过硅藻土过滤,并且用etoac冲洗滤饼。分离得到的各层,并且用饱和nacl水溶液洗涤有机相两次,经mgso4干燥,过滤,并且真空浓缩。向所得残留物中加入水(500ml),在rt搅拌16小时,并且过滤所得固体。用
h2o洗涤收集的固体,并且真空干燥16小时,得到所需化合物(70g,54%),为棕色固体。es/ms(m/z)(
35
cl/
37
cl)267/269[m 1]
[0106]
制备例4
[0107]
反式-5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-2,4-二甲氧基-嘧啶
[0108][0109]
将4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-哒嗪(1.97g,7.39mmol)、外消旋-反式-2-[2-异丙基环丙基]-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(3.32g,15.8mmol)、双(二-叔丁基(4-二甲基氨基苯基)膦)二氯钯(ii)(1.35g,1.85mmol)、1,4-二烷(37ml)和1m na2co3水溶液(18ml,18mmol)合并。用n2吹扫反应容器,并且加热至90℃达18小时。冷却至rt,用etoac(150ml)稀释,并且分离各层。依次用1m na2co3水溶液、饱和nacl水溶液洗涤有机层,经mgso4干燥,过滤,并且真空浓缩滤液。通过硅胶色谱法纯化所得残留物,用60-100%etoac/dcm梯度洗脱,在蒸发色谱级分后得到标题化合物,为异构体的基本上外消旋的混合物(1.48g,64%)。
[0110]
将异构体经sfc纯化(柱:cellulose-4,4.6
×
150mm;40%meoh/co2等度;流速:5ml/分钟,uv 250nm),得到647mg异构体1:tr=2.56分钟和647mg异构体2:tr=3.75分钟。es/ms(m/z):315(m h)。
[0111]
制备例5
[0112]
(2s)-2-氯-3-甲基-丁酸
[0113][0114]
将l-缬氨酸(286g,2.44mol)溶液和5m hcl水溶液(3.25l,16.3mol)在0℃冷却。历经2小时滴加4m nano2水溶液(1l,4mol),保持内部温度低于5℃,将反应搅拌2小时,同时温至rt,并且在rt搅拌另外16小时。历经30分钟分批加入na2co3(242g,2.28mol)。用mtbe(3
×
1000ml)萃取得到的溶液,用饱和nacl水溶液(500ml)洗涤合并的有机萃取物,经mgso4干燥有机萃取物,并且真空浓缩。通过真空蒸馏(15mbar/140℃)纯化所得残留物,得到标题化合物(248g,68%),为油状物。1h nmr(400mhz,cdcl3)δ:1.1(dt,j=6.6,2.6hz,6h),2.34-2.42(m,1h),4.19-4.23(m,1h),10.0-12.0(br-s,1h)。
[0115]
制备例6
[0116]
(2s)-2-氯-3-甲基-丁-1-醇
[0117][0118]
将(2s)-2-氯-3-甲基-丁酸(137g,1mol)在2-甲基四氢呋喃(500ml)中的溶液冷却至0℃,历经2.5小时滴加入2.3m lah在甲基四氢呋喃中的溶液(480ml,1.1mol),保持内部温度低于10℃。温至rt并且将混合物在rt搅拌1小时,并且在50℃搅拌1小时,将反应冷却至
0℃,并且依次缓慢加入h2o(1.48ml)、15%naoh水溶液(1.48ml)和h2o(4.46ml)。使混合物温至rt,通过硅藻土床过滤,并且真空蒸发溶剂,得到标题化合物(110g,81%),为无色油状物。1h nmr(400mhz,cdcl3)δ:0.97-1.1(m,6h),1.94-2.18(m,1h),2.60-3.09(br-s,1h),3.71-3.78(m,1h),3.80-3.85(m,1h),3.90-3.96(m,1h)。
[0119]
制备例7
[0120]
(2r)-2-异丙基环氧乙烷
[0121][0122]
将koh(195g,3.48mol)在h2o(195ml)中的溶液冷却至0℃,并且历经20分钟加入纯的(2s)-2-氯-3-甲基-丁-1-醇(110g,801mmol),同时保持内部温度低于5℃。使反应混合物温至rt。通过在100mbar真空蒸馏从23℃升温至50℃来纯化反应混合物,得到标题化合物(47g,64%),为无色油状物。1h nmr(400mhz,cdcl3)δ:0.98(d,j=6.9hz,3h),1.05(d,j=6.9hz,3h),1.51(o,j=6.9hz,1h),2.52-2.54(m,1h),2.70-2.75(m,2h)。
[0123]
制备例8
[0124]
(1s,2r)-2-异丙基环丙烷甲酸乙酯
[0125][0126]
历经25分钟将2.5m nbuli在己烷中的溶液(310ml,780mmol)滴加至在冰/水浴(内部温度:8℃)中冷却的2-二乙氧基磷酰基乙酸乙酯(153ml,772mmol)在1,4-二烷(870ml)中的溶液中。温至rt并且搅拌40分钟。将溶液通过套管转移至3l压力容器中,并且加入在1,4-二烷(180ml)中的(2r)-2-异丙基环氧乙烷(70g,772mmol)。在150℃和50psi压力下搅拌反应混合物14小时。将反应混合物冷却至rt,并且加入h2o(700ml)。分离各层,并且用mtbe(2
×
500ml)反萃取水相。合并有机相,用饱和nacl水溶液(2
×
350ml)洗涤,经mgso4干燥,并且真空浓缩,得到粗标题化合物(107.7g,>99%),为黄色油状物,其适于随后使用而无需进一步纯化。gc-ms(m/z):156(m )。
[0127]
制备例9
[0128]
(1s,2r)-2-异丙基环丙烷甲酸
[0129][0130]
将(1s,2r)-2-异丙基环丙烷甲酸乙酯(107.7g,482.6mmol)在1,4-二烷(800ml)中的溶液与25%naoh水溶液(800ml)的混合物在100℃搅拌7小时。将混合物冷却至rt,加入水(300ml),用mtbe(2
×
500ml)萃取,并且弃去有机相。用37%hcl水溶液(约500ml)酸化水相直到ph为~1-2。
[0131]
用mtbe(2
×
600ml)萃取酸化的含水混合物,用饱和nacl水溶液洗涤有机层,经mgso4干燥,并且真空浓缩,得到标题化合物(54.2g,75%),为琥珀色油状物。1h nmr(400mhz,cdcl3)δ:0.81-0.86(m,1h),1.01(dd,j=5.9,3.7hz,6h),1.04-1.13(m,1h),1.20-1.24(m,1h),1.28-1.37(m,1h),1.39-1.44(m,1h)。
[0132]
制备例10
[0133]
(1s,2r)-2-异丙基环丙烷甲酸(1,3-二氧代异吲哚啉-2-基)酯
[0134][0135]
将(1s,2r)-2-异丙基环丙烷甲酸(54.2g,359mmol)、2-羟基异吲哚啉-1,3-二酮(59.8g,359mmol)和dmap(4.44g,35.9mmol)在dcm(690ml)中的混悬液在0℃搅拌。滴加n,n
’‑
二异丙基碳二亚胺(50.4g,395mmol),温至rt,并且将得到的反应混合物在rt搅拌2小时。加入h2o(600ml),并且分离各相。用dcm(2
×
300ml)萃取水相,合并有机相,经mgso4干燥,过滤并且蒸发。通过硅胶色谱法纯化所得固体残留物,用100%dcm洗脱,蒸发色谱级分后,得到标题化合物(96g,88%),为浅黄色固体。es/ms(m/z):274(m 1)。
[0136]
制备例11
[0137]
2-[(1s,2s)-2-异丙基环丙基]-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷
[0138][0139]
使氮气流通过(1s,2s)-2-异丙基环丙烷甲酸(1,3-二氧代异吲哚啉-2-基)酯(96g,316mmol)和二(频哪醇基)二硼(160g,630mmol)在etoac(480ml)中的溶液15分钟。将混合物在85℃搅拌,并且历经10分钟滴加异烟酸乙酯(24.38g,157mmol)。将所得混合物在85℃搅拌16小时。冷却所得混悬液,过滤并且弃去固体,并且减压蒸发棕色滤液。将粗残留物经硅胶填料过滤,用2%etoac/己烷洗脱。从滤液中除去溶剂,并且用硅胶色谱法对所得残留物进行再纯化,用3%etoac/己烷洗脱,从色谱级分中除去溶剂后,得到标题化合物(25.3g,38%),为无色油状物。1h nmr(400mhz,cdcl3):-0.33_-0.38(m,1h),0.42-0.47(m,1h),0.63-0.67(m,1h),0.75-0.82(m,1h),0.89-1.02(m,1h),0.98(m,6h),1.24(s,12h)。
[0140]
制备例12
[0141]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-2,4-二甲氧基-嘧啶
[0142][0143]
将4-氯-6-(2,4-二甲氧基嘧啶-5-基)-3-甲基-哒嗪(19.1g,70.6mmol)、k3po4(45.9g,212mmol)、1,4-二烷(300ml)和h2o(75ml)混合,并且用n2使混合物脱气10分钟。加入[1,1
’‑
双(二苯基膦基)二茂铁]二氯钯(ii)(7.98g,10.6mmol)。用氮气鼓泡另外2分钟,并且一次性加入2-[(1s,2s)-2-异丙基环丙基]-4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(22.2g,106mmol)。将所得混合物在80℃搅拌16小时。将反应冷却至rt,加入h2o,并且用etoac萃取。分离有机层,经无水mgso4干燥,并且减压浓缩。通过硅胶色谱纯化得到的残留物,用85%己烷/etoac洗脱,从色谱馏分中除去溶剂后,分离出标题化合物(21.5g,92%),为琥珀色油状物。将油状物在rt静置固化成灰白色固体。es/ms(m/z):315(m 1)。
[0144]
实施例1
[0145]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮
[0146][0147]
将反式-5-[5-[2-异丙基环丙基]-6-甲基-哒嗪-3-基]-2,4-二甲氧基-嘧啶异构体1(628mg,2.00mmol)溶解在meoh(3ml)中。加入1m hcl水溶液(5ml),并且加热至70℃达3小时。冷却至rt,将反应混合物上样于meoh洗涤的scx柱(20g,silicycle siliabond tosic acid),用meoh(140ml)洗涤scx柱,并且用2m nh3/meoh(140ml)洗脱所需产物。浓缩nh3/meoh级分,得到标题化合物,为奶油色固体(546mg,95%)。es/ms(m/z):287(m h)。1h nmr(d
6-dmso)δ:0.99(m,9h),1.24(m,1h),1.81(m,1h),2.72(s,3h),7.67(s,1h),8.23(s,1h),11.43(bs,2h)。
[0148]
结晶形式的x-射线粉末衍射
[0149]
将游离碱化合物溶于甲醇中,在50℃搅拌1小时,并且使其冷却至环境温度,在此其从溶液中结晶。然后通过真空过滤分离固体,并且在70℃真空短暂干燥。
[0150]
结晶固体的xrd图在bruker d4 deimer x-射线粉末衍射仪上获得,该衍射仪装备有cuka源)和vantec检测器,在35kv和50ma操作。样品在4和40
°
(2θ)之间扫描,具有0.008
°
(2θ)的步长并且扫描速率为0.5秒/步,并且具有0.6mm的发散度、5.28的固定抗散射和9.5mm的检测器狭缝。将干燥粉末装在石英样品架上,并且使用载玻片获得光滑表面。在环境温度和相对湿度下收集晶体形式衍射图。在晶体学领域中众所周知,对于任何给定的晶体形式,衍射峰的相对强度可能由于由诸如晶体形态和习性等因素导致的优选取向而变化。当存在优选取向的影响时,峰强度改变,但是多晶型物的特征峰位置不变。参见例如美国药典#23,国家处方集#18,第1843-1844页,1995。此外,在晶体学领域中也众所周知,对于任何给定的晶体形式,角峰位置可以稍微变化。例如,峰位置可能由于分析样品的温度或湿度的变化、样品位移或内标的存在或不存在而偏移。在本情况下,
±
0.2(2θ)的峰位置变化将考虑这些潜在变化,而不妨碍所指示的晶体形式的明确鉴定。晶体形式的确认可以基于区别峰(以
°
2θ为单位)的任何独特组合,通常是更显著的峰。基于在8.853和26.774度(2θ)处的nist 675标准峰调整在环境温度和相对湿度下采集的晶体形式衍射图。
[0151]
13
c固态nmr(
13
cssnmr)
[0152]
13
c交叉极化/魔角旋转nmr(固态nmr或ssnmr)谱是使用bruker avance ii 400mhz nmr光谱仪获得的,该光谱仪在100.622mhz碳频率和400.131mhz质子频率下操作,并且装有bruker 4mm双共振探针。toss边带抑制与采用spinal64去耦和ramp 100形h-核cp脉冲的交叉极化一起使用。采集参数如下:90
°
质子r.f.脉冲宽度为2.6μs,接触时间为2.0ms,脉冲重复时间为3s,mas频率为10khz,谱宽为30khz,采集时间为34ms,以及扫描次数为18,661次。化学位移在单独的试验中参照金刚烷(δ=29.5ppm)。
[0153]
制备例13
[0154]
结晶形式1,5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,
4-二酮(无水)
[0155][0156]
将5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-2,4-二甲氧基-嘧啶(21.5g,65.6mmol)和1m hcl水溶液(165ml,135mmol)的混悬液在45℃搅拌16小时。冷却至rt,并且用mtbe萃取。弃去有机相,并且向水相中加入2m k2hpo4水溶液直至ph~6(约150ml)。将所得混合物在rt搅拌16小时。过滤并且收集所得固体,用水洗涤并且在真空烘箱中在45℃干燥16小时,得到标题化合物(14.3g,76%),为白色固体。es/ms(m/z):287(m 1)。
[0157]
制备的5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮无水结晶形式(形式1)的样品的特征在于使用cuka辐射的xrd图具有如下表1中所述的衍射峰(2θ值)。具体地,所述图包含在4.7
°
处的峰与选自11.7
°
、15.8
°
和16.0
°
的一个或多个峰的组合;衍射角的允许误差为0.2度。
[0158]
表1:5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮无水结晶形式(形式1)的x-射线粉末衍射峰。
[0159][0160]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮无水结晶形式(形式1)代表性
13
c ssnmr共振包括:16.6、18.3、20.7、22.1、23.9、25.4、26.6、27.6、28.1、28.9、30.2、31.2、32.2、38.6、39.7、41.2、43.1、109.3、111.0、112.0、124.4、128.2、130.7、145.1、146.3、150.1、151.0、153.4、156.7、157.6、158.6、159.3、160.7、163.4、165.9、167.0、169.4和170.2ppm(分别为
±
0.2ppm)。
[0161]
制备例14
[0162]
结晶形式2,5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮(去溶剂化物/无水)
[0163]
在3500l hastelloy反应器中,于rt将5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮(15.4kg,53.8mol)加入到水(770l)中。将所得混悬液在rt搅拌19小时。过滤并且收集所得固体,用水洗涤并且在真空烘箱中于40℃干燥3天,得到标
题化合物(13.8kg,89%),为白色固体。hlpc纯度:99.8%。
[0164]
观察到从5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮去溶剂化结晶形式(形式2)的样品制备的相关结晶形式家族。一个家族成员的特征在于使用cuka辐射的xrd图具有下表2中所述的衍射峰(2-θ值)。
[0165]
表2:5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮去溶剂化结晶形式(形式2)的x-射线粉末衍射峰。
[0166][0167]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮去溶剂化(无水)结晶形式(形式2)的代表性
13
c ssnmr共振包括:17.2、18.9、21.1、22.5、24.4、26.7、27.8、28.7、29.7、39.7、41.3、45.5、50.1、110.7、111.7、112.4、122.0、125.0、126.1、129.1、143.6、144.8、146.2、147.2、149.9、151.1、153.0、155.5、157.2、159.0、160.6、161.4、162.7、164.8、167.0、168.3和170.4ppm(分别为
±
0.2ppm)。
[0168]
其它家族成员的特征在于使用cuka辐射的xrd图谱,其具有大于4.3
°
并且至多5.5
°
的衍射峰(2θ值),与4.7
°
、6.4
°
、9.3
°
、11.0
°
、11.6
°
、18.4
°
和27.9
°
处的一个或多个峰组合,衍射角的允许误差为0.2度。
[0169]
制备例15
[0170]
结晶形式3,5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮(半水合物)
[0171]
在rt,将5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-2,4-二甲氧基-嘧啶(316g,0.92mol)加入到5l烧瓶中的1m hcl水溶液(2.6l,2.6mol)中。将所得混悬液加热至45℃并且在该温度搅拌16小时。冷却至rt,并且加入2m k2hpo4水溶液直至ph~6。将所得混合物在rt搅拌1小时。过滤并且收集所得固体,在真空烘箱中于40℃干燥3天,得到标题化合物(250g,92%),为白色固体。hlpc纯度:99%。
[0172]
制备的5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮半水合物结晶形式(形式3)的样品的特征在于使用cuka辐射的xrd图,其具有如下表3中所述的衍射峰(2θ值)。具体地,该图包含在5.0
°
的峰与选自16.4
°
、17.8
°
和26.6
°
的一个或多个峰的组合;衍射角的允许误差为0.2度。
[0173]
表3:5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮半水合物结晶形式(形式3)的x-射线粉末衍射峰。
基]-1h-嘧啶-2,4-二酮对抗cd73酶活性。将含有2μm单磷酸腺苷(sigma#01930)、10mm tris ph 7.5、100mm nacl、0.01%bsa、0.2mm辛基葡糖苷和50pm cd73蛋白的反应混合物(20μl)加入384孔板(nunc#264573)中。rt温育30分钟后,加入20μl含2%甲酸和10μm 13
c5-腺苷(用
13
c5标记的核糖)(cambridge isotope laboratories-#clm-3678-0)的终止溶液终止反应,然后加入40μl dh2o。腺苷-和腺苷核糖-13
c5(内标)水平使用如上所述的质谱测定。使用信号比(腺苷峰积分/腺苷内标峰积分)来定量每个反应。通过使用方程{%抑制=100x[1-(x-min)/(max-min)]}计算抑制百分比,其中x等于孔信号比,max等于dmso对照的信号比中值,并且min等于在存在》10x ic
50
的已知竞争性抑制剂的情况下的酶活性信号比。为了筛选目的,在50μm于1%dmso中测试各化合物。通过测试10个浓度为0.0025至50μm的各化合物(使用1:3稀释方案)测定5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的ic
50
。
[0186]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的ic
50
为0.028μm。
[0187]
小鼠cd73机制测定
[0188]
该测定的目的是评价5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮对小鼠cd73酶活性的抑制。该测定如上文关于人cd73生化测定所述进行,不同之处在于使用3μm amp和50pm小鼠cd73酶。
[0189]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的ic
50
是0.175μm。
[0190]
calu6人细胞测定
[0191]
该测定的目的是在基于细胞的测试中测试5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮对抗cd73。将calu6细胞(1500个细胞/孔)于96孔涂覆poly-d-赖氨酸的板(bd#356640)中生长,所述板包含100μl培养基(mem(gibco#11095-072) 1%丙酮酸钠(gibco#11360-070) 1%neaa(gibco#11140-050) 10%fbs(hyclone#sh30071)。将板在rt温育30分钟,然后在37℃/5%co2下温育过夜。用测定缓冲液(10mm tris-hcl ph 7.2、10mm d-葡萄糖、1mm kcl、125mm nacl、2mm mgcl2)(90μl/孔)洗涤细胞两次。然后,向每孔中加入90μl测定缓冲液,接着每孔加入10μl amp和化合物预混物(50μm amp,在1%dmso中的5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的不同浓度)。将板在室温温育60分钟。然后,除去每孔10μl上清液,并且加入到新的板中,随后加入20μl终止液(2%甲酸,1.2μm腺苷核糖-13
c5(cambridge isotope laboratories-#clm-3678-0)和90μl ddh2o用于质谱分析。通过使用质谱(agilent rapidfire)测定腺苷-和腺苷核糖-13
c5(内标)水平,如上文对人cd73生化测定所述。抑制百分比也如上文所述计算。
[0192]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的ic
50
为0.0073um(实施例2的化合物)。
[0193]
离体靶抑制测定
[0194]
该测定的目的是在基于离体的测定中测试5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮在小鼠血液中对抗鼠cd73。在肿瘤达到约400mm3后,给动物(6只/组)口服给予在20%hpbcd(2-羟丙基-β-环糊精)、ph 2中配制的每种化合物。
处理后,将血液收集到肝素管中,并且用于
13
c10-15
n5-amp向标记腺苷、肌苷和次黄嘌呤的转化的离体分析,如使用通过从口服给药而接受化合物处理的动物收集的全血进行的离体测定所述。
[0195]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮在来自用不同剂量的化合物治疗的动物的小鼠全血中抑制amp向腺苷、肌苷和次黄嘌呤的转化,如表4所示。
[0196][0197][0198]
*:统计学显著。
[0199]
5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的ic
50
为0.0073um。
[0200]
基于calu6肿瘤的人cd73体内靶向抑制的测定
[0201]
该测定的目的是在体内靶向抑制测定中测试5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮在衍生自人癌症calu6细胞的异种移植肿瘤中对抗人cd73。calu6细胞(atcc)在补充有10%胎牛血清的hbss培养基中生长。用胰蛋白酶收获近汇合的细胞,并且用缺乏血清的生长培养基冲洗两次。通过在裸鼠(harlon laboratory)的后胁注射在hbss和matrigel的1:1混合物(bd biosciences,franklin lakes,nj)中的5
×
106来引发皮下肿瘤的生长。当平均肿瘤体积达到约400-500mm3时,根据肿瘤大小和体重随机排列动物,并且如所示放入它们各自的治疗组中。处理后,收集肿瘤样品(每个50-80mg)并且在1ml含有如下所述内标的冰冷提取缓冲液中处理。
[0202]
将箔条和液体n2加入到研钵中以预冷却。将肿瘤组织滴在箔条上,并且加入液体n2。将另一箔条置于肿瘤组织的顶部,并且用杵锤击直至肿瘤完全平整。将50至100mg肿瘤组织置于试管中(fishers scientific,cat#02-681-302)并且置于干冰上。向管中加入一个金属珠(qiagen cat.no.69989)和1ml 80%的甲醇,其含有内标
13c5-腺苷、
13c5-amp、
15n5-gtp、
15n4-肌苷5
’‑
单磷酸和
13
c-15
n-次黄嘌呤(cambridge isotope lab和cayman chemical),并且将样品贮存于-80℃直到用于lc/ms分析。
[0203]
将血液也收集到肝素管中,并且用于
13c5-15
n5-amp向标记腺苷、肌苷和次黄嘌呤转化的离体分析,如使用从通过口服给药接受化合物处理的动物收集的全血的离体测定所
述。
[0204]
如表5所示,5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮在用不同剂量的化合物治疗的calu6肿瘤中抑制amp向腺苷的转化。
[0205][0206]
*:统计学显著。
[0207]
基于lc/ms的用于测量人血清中cd73活性的测定
[0208]
将新鲜的正常人血液在室温1,500g离心15分钟,并且收集含有血清的上层级分。将收集的血清(25μl/孔)转移到96-深孔板(dwp,analytical sales&services inc.cat#968820)中,该板含有多种浓度的5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮和固定浓度的左旋咪唑(1,500μm)。在冰上温育60分钟后,将
13
c5-15
n5-amp(50um)加入到板的每个孔中,并且将板在室温温育15分钟。然后将板置于干冰上,加入200ul/孔的17.3tca,随后用板振摇器(qiagen)以26fps振摇3分钟。然后将板在4℃以2940g离心20分钟。离心后,将每孔100ul/孔的上清液转移到新的96深孔板中,并且与冰上18.4ul/孔的2.5m na2co3混合,随后加入200ul含有内标(is:
13
c5-amp、
13
c5-腺苷、
13
c5-次黄嘌呤和
15
n4-肌苷)的提取溶液。在4℃以2940g进一步离心20分钟后,将200ul/孔的上清液用于如上所述通过lc/ms分析
13
c10-15
n5-腺苷、
13
c10-15
n5-肌苷和
15
n5-次黄嘌呤。对于ec50计算,用来自计算机模型或试验的未结合级分(%)校正血清中5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮的浓度。
[0209]
表6包含5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮在人血清中抑制cd73活性的数据。
[0210][0211]
体内模型
[0212]
如果需要,5-[5-[(1s,2r)-2-异丙基环丙基]-6-甲基-哒嗪-3-基]-1h-嘧啶-2,4-二酮或其药学上可接受的盐的cd73抑制作用的临床前建模可以例如根据本领域中所述的方法进行,例如rongvaux a等人,annual rev.immunology 2013;31:635-74,以及其中引用的文献;sanmamed mf等人,annals of oncology 2016;27:1190-1198,以及其中引用的文献。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。