工程化的调节t细胞的产生
1.相关申请的交叉引用
2.本技术要求于2019年11月8日提交的美国临时申请62/933,252的优先权,其内容通过引用整体并入本文。
背景技术:
3.健康的免疫系统是平衡的。参与适应性免疫的细胞包括b和t淋巴细胞。有两种一般类型的t淋巴细胞——效应t(teff)细胞和调节性t(treg)细胞。teff细胞包括cd4
t辅助细胞和cd8
细胞毒性t细胞。teff细胞在抗原挑战后的细胞介导免疫中起核心作用。teff细胞和其他免疫细胞的关键调节因子是treg细胞,它可以防止过度的免疫反应和自身免疫(参见例如,romano et al.,front immunol.(2019)10,art.43)。
4.一些treg在胸腺中产生;它们被称为天然treg(ntreg)或胸腺treg(ttreg)。其他tregs是在遇到抗原后在周围产生或在细胞培养物中产生,其称为诱导treg(itreg)或适应性treg。treg通过涉及特定细胞表面受体的细胞间接触和抑制性细胞因子如il-10、tgf-β和il-35的分泌主动控制其他免疫细胞的增殖和激活,包括诱导耐受(dominguez-villar and hafler,nat immunol.(2018)19:665-73)。诱导耐受失败可导致自身免疫和慢性炎症。耐受丧失可能是由treg功能缺陷或treg数量不足或者teff无反应或过度激活所致(sadlon et al.,clin transl immunol.(2018)7:e1011,doi:10-1002/cti2.1011)。
5.近年来,人们对使用treg治疗疾病产生了浓厚的兴趣。已经探索了许多方法,包括过继细胞疗法,以提高treg的数量和功能,以治疗自身免疫疾病。treg转移,其递送激活的和扩增的treg群,已在患有自身免疫疾病,如i型糖尿病、皮肤红斑狼疮和克罗恩病的患者以及在器官移植中进行了测试(dominguez-villar,supra;safinia et al.,front immunol.(2018)9:354)。
6.目前,用于细胞疗法的treg的唯一来源是成人或青少年的原血(例如,全血或单采产品)和组织(例如,胸腺)。从这些来源中分离treg是侵入性的且耗时,并且仅产生少量treg。此外,从这些样品中获得的treg本质上是多克隆的,并且可以在其潜在的免疫抑制反应中引入变异性。还有证据表明,仅仅增加treg的数量可能不足以控制疾病(mcgovern et al.,front immunol.(2017)8,art.1517)。具有抗原特异性部分的工程化单克隆treg,如car或工程化tcr,可能会在自身免疫活性或器官移植位点增强免疫调节反应。仍然需要有效地获得大量基因工程化的单克隆treg细胞。
7.发明概述
8.本公开提供了用于促进干细胞,包括诱导全能干细胞(ipsc)和祖细胞分化成调节t细胞的方法和组合物。在优选的实施方案中,制备工程化的调节t细胞用于过继细胞疗法。
9.在一个方面,本公开提供了在基因组中包含异源序列的基因工程化的哺乳动物细胞(例如,人细胞),其中该异源序列包含编码谱系确定因子(本文也称为谱系诱导因子)的转基因,和其中谱系定型因子促进细胞分化为cd4
调节t细胞(treg)或促进细胞维持为cd4
treg。在一些实施方案中,异源序列被整合到工程化的细胞基因组中的安全港位点(例如,
aavs1基因座)。在其他实施方案中,异源序列被整合到t细胞特异性基因座中,即,含有在t细胞,如treg中特异性表达的基因的基因座(例如,foxp3位点和helios位点);在这些实施方案中,转基因可以是在基因座中的转录调控元件的控制下。
10.在另一个方面,本公开提供了制造基因工程化的哺乳动物细胞的方法,其包括:使哺乳动物细胞与包含下项的核酸构建体接触:(i)异源序列和(ii)第一同源区(hr)和异源序列侧翼的第二hr,其中异源序列包含转基因,第一和第二hr分别与在哺乳动物细胞中的t细胞特异性基因座或基因组安全港中的第一基因组区(gr)和第二gr同源;以及在允许在t细胞特异性基因座或基因组安全港中的第一和第二gr之间的异源序列整合的条件下培养细胞。在一些实施方案中,异源序列整合由锌指核酸酶或切口酶(zfn)、转录激活因子样效应结构域核酸酶或切口酶(talen)、大范围核酸酶、整合酶、重组酶、转座酶或crispr/cas系统促进。在一些实施方案中,核酸构建体是慢病毒构建体、腺病毒构建体、腺相关病毒构建体、质粒、dna构建体或rna构建体。
11.在一些实施方案中,转基因包含附加多肽的编码序列,其中谱系定型因子的编码序列和附加多肽的编码序列由自切割肽的框内编码序列或由内部核糖体进入位点(ires)分离。在特定实施方案中,附加多肽是另一种谱系定型因子、治疗性蛋白或嵌合抗原受体(car)。
12.在一些实施方案中,异源序列整合到t细胞特异性基因座中的外显子中,并且包含:紧邻转基因上游的内部核糖体进入位点(ires);或紧邻转基因上游并与转基因框内的自切割肽的第二编码序列。在进一步的实施方案中,异源序列进一步包含紧邻ires或自切割肽的第二编码序列上游的核苷酸序列,该核苷酸序列包含整合位点下游的t细胞特异性基因座的所有外显子序列,使得t细胞特异性基因座仍然能够表达完整的t细胞特异性基因产物。在特定实施方案中,t细胞特异性基因座是t细胞受体α恒定(trac)基因座,并且异源序列任选整合到trac基因座的外显子1、2或3中。
13.在一些实施方案中,转基因编码foxp3、helios或thpok。在进一步的实施方案中,转基因包含foxp3的编码序列和thpok的编码序列,其中这两个编码序列是框内的并且由自切割肽的框内的编码序列分离。
14.在一些实施方案中,细胞是人细胞。在进一步的实施方案中,细胞是干细胞或祖细胞,任选地选自胚胎干细胞、诱导全能干细胞、中胚层干细胞、间充质干细胞、造血干细胞、淋巴祖细胞或祖t细胞。在一些实施方案中,细胞从t细胞(例如,treg、cd4
t细胞或cd8
t细胞)重编程。在一些实施方案中,工程化的细胞是treg。
15.在一些实施方案中,本公开提供了产生工程化treg的方法,该方法包括:在包含下项的组织培养基中培养本文工程化的细胞或祖细胞:(i)低il-2剂量,(ii)il-7ra(cd27)的信号传导抑制剂(例如,抗体),(iii)ccr7信号传导的抑制剂(例如,抗体)。在一些实施方案中,本公开提供了产生工程化的treg的方法,该方法包括:共培养本文的工程化的干细胞或祖细胞与ms5-dll1/4基质细胞;op9或op9-dll1基质细胞;或epcam-cd56
基质细胞。本公开还提供通过这些方法获得的treg细胞。
16.在一些实施方案中,工程化的细胞进一步包含基因的无效突变,该基因选自ii类主要组织相容性复合物反式激活因子(ciita)基因、hla i类或ii类基因、与抗原加工相关的转运蛋白、次要组织相容性抗原基因和β2微球蛋白(b2m)基因。
17.在一些实施方案中,工程化细胞进一步包含自杀基因,其任选地选自hsv-tk基因、胞嘧啶脱氨酶基因、硝基还原酶基因、细胞色素p450基因或caspase-9基因。
18.本公开进一步提供了治疗需要免疫抑制的患者(例如,人患者)的方法,其包括向患者施用本文提供的工程化的细胞(例如,工程化的treg)。还提供了本文的工程化的细胞在制备用于治疗需要免疫抑制的患者(例如,人患者)的药物中的用途,以及本文的工程化细胞用于治疗需要免疫抑制的患者(例如人患者)。在一些实施方案中,患者患有自身免疫疾病或已经接受或将接受组织移植。
19.本发明的其他特点、目的和优点在下面的详细描述中是显而易见的。然而,应该理解的是,详细描述虽然指示了本发明的实施方案和方面,但仅以阐释方式给出而不是限制性的。根据详细描述,本领域技术人员将清楚本发明范围内的各种变化和修饰。
附图说明
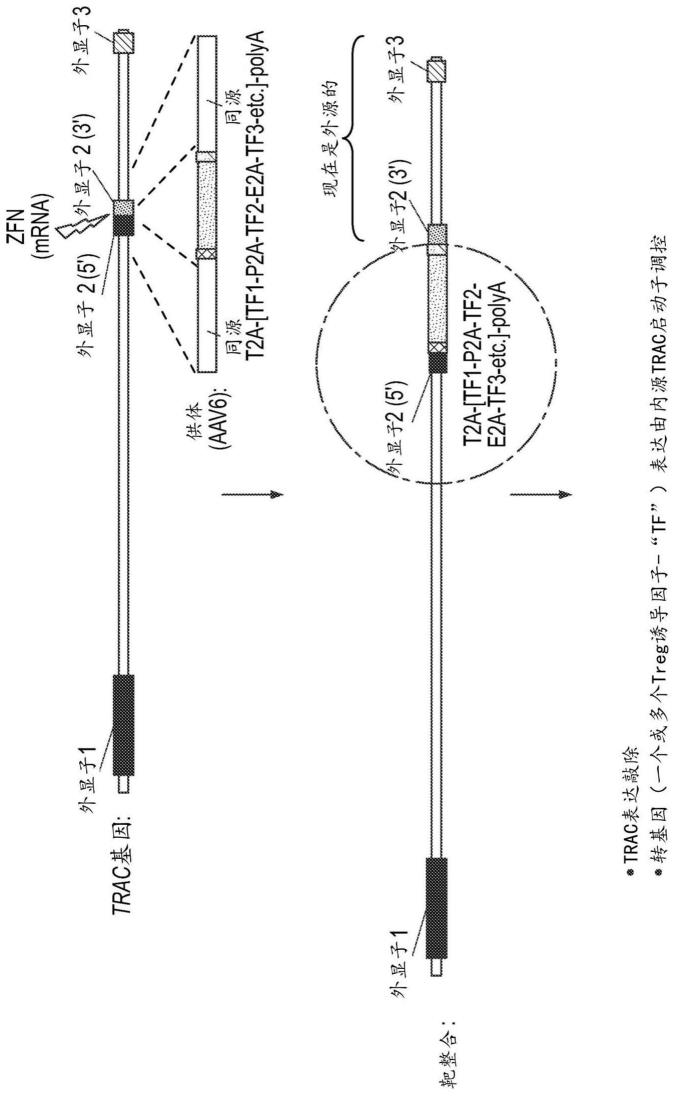
20.图1是描述将编码一种或多种treg定型(或诱导)因子(“tf”)的转基因整合到人trac基因的外显子2中的基因组编辑方法的示意图。由引入的mrna产生的锌指核酸酶(zfn)在外显子2的特定位点(闪电)发生双链断裂。由腺相关病毒(aav)6载体引入的供体序列自5'至3'含有:同源区1;自切割肽t2a的编码序列;用于融合第一tf、自切割肽p2a、第二tf2、自切割肽e2a和第三tf3的编码序列;多聚腺苷酸化(polya)信号序列;和同源区2。同源区与zfn切割位点侧翼的基因组区同源。整合位点上游的trac外显子2部分、t2a编码序列和tf编码序列彼此在框内。在这种方法下,由于转基因整合,敲除trac蛋白的表达。整合序列的表达通过内源tcrα链启动子的调节。
21.图2是描述类似于图1中描述的基因组编辑方法的示意图,但在此,异源序列包含部分trac cdna,其涵盖整合位点下游的trac外显子序列(即外显子2序列3'到整合位点和外显子3序列)。该部分trac cdna直接置于t2a编码序列的上游并与t2a编码序列在框内,使得工程化的基因座表达完整的tcrα链和在内源tcrα链启动子下的tf。
22.图3是描述整合编码一个或多个定型因子的转基因的另一种基因组编辑方法的示意图。在这种方法中,整合转基因到基因组安全港中。在该图中,插入转基因到人aavs1基因座的内含子1中,并与强力霉素(dox)诱导启动子可操作地连接。sa:剪接受体。图2a:自切割肽2a的编码序列。puror:嘌呤霉素抗性基因。ti:靶向的整合。
23.图4是一组图表,显示了从使用图3中概述的示意图编辑的细胞中生成的数据。转基因编码绿色荧光蛋白(gfp)。puro:嘌呤霉素。dox:强力霉素。
24.图5是描述基因组编辑方法的示意图,其中将编码一种或多种定型因子的转基因整合到人aavs1基因的内含子1中。整合到基因组中的异源序列包括car编码序列。一旦treg分化完成,就切除编码定型因子的转基因(放置在两个loxp位点之间),在整合位点仅留下car表达盒。
25.图6是描述将编码一种或多种定型因子的转基因整合到人trac基因的外显子2中的基因组编辑方法的示意图。在这种方法中,整合到基因组中的异源序列包括car编码序列。一旦treg分化完成,就切除编码定型因子的转基因(放置在两个loxp位点之间),在整合位点仅留下car表达盒。
26.图7是描述用于将具有单重排tcr的成熟treg重编程为诱导全能干细胞(ipsc)的
过程的示意图。扩增后,ipsc重新分化回treg表型。这里的tcr靶向非同种异体抗原的抗原。
27.图8是描绘ipsc分化成treg的过程的示意图。hsc:造血干细胞。单阳性:cd4
或cd8
。双阳性:cd4
cd8
。
28.图9是一组细胞分选图,展示向组织培养基中引入针对il-7受体(il-7ra)的α单元的抗体会使ipsc衍生的祖t细胞分化不形成cd8单阳性细胞(左上象限)以形成cd4单阳性细胞(右下象限)。将抗体以三种浓度(低、中和高)添加到组织培养基中。这种效果显示在两个独立的实验(expt.#1和expt.#2)中。
29.图10是描绘用于将ipsc分化成treg的多个过程的示意图。细胞在淋巴细胞分化涂层材料(不依赖于饲养层)上或与op9基质细胞或op9-dll1基质细胞(表达notch配体,delta样1的op9细胞)基质细胞(依赖于饲养层)培养。然后如图8所示进一步培养细胞以促进其分化成treg。在另一条路径中,三维胚胎中胚层类器官(emo)是通过与mss-dll1/4或epcam-cd56
基质细胞共培养ipsc而形成的;在emo的造血诱导后,人工胸腺类器官(ato)形成,其被诱导产生成熟的treg,其tcr库更类似于胸腺选择的treg。
30.图11是描述整合crispr活化(crispra)或抑制(crispri)文库的基因组编辑方法的示意图,该文库包括分别与vph活化域或krab抑制域融合的死cas9(dcas9)。在该图中,整合文库(转基因)到人aavs1基因的内含子1中。
31.图12是一组图表,比较衍生自幼稚调节、cd4
和cd8
t细胞(统称为tipsc)的ipsc与衍生自cd34
细胞的ipsc之间产生t细胞的能力。图a显示了在tipsc和cd34衍生的ipsc分化期间共表达cd3和tcrαβ的活/单细胞的百分比。图b是一组具有代表性的流式细胞术图,描绘了在将t细胞与ipsc区分开来时cd3和tcrαβ的表达。还检查了来自每个ipsc系的cd3
tcrαβ
细胞(图c)以及来自活/单细胞(图d)的细胞类型和分布。cd4sp:cd4单阳性。cd8sp:cd8单阳性。dn:双阴性(cd4-cd8-)。dp:双阳性(cd4
cd8
)。统计显著性由未配对t检测和welch校正确定。星号表示统计显著性。
32.图13a是一组流式细胞术图,显示了在图2中阐释的方法中衍生自在trac基因座的外显子2处编辑的ipsc系的t细胞中foxp3和抗hla-a2嵌合抗原受体(car)的表达。转基因是foxp3/helios/car、foxp3/car、foxp3或gfp。
33.图13b是显示图13a研究中细胞的细胞因子分泌分析的图。
34.详细说明
35.全能干细胞(psc)可以无限扩增并产生人体内的任何细胞类型。psc(例如人胚胎干细胞和诱导的全能干细胞)代表了用于产生大量用于治疗应用的分化细胞的理想起始来源。本公开提供了从如诱导的psc(ipsc)的psc产生treg细胞的方法。本公开还包括从多能细胞如中胚层祖细胞、造血干细胞或淋巴祖细胞产生treg细胞的方法。与全能细胞相比,多能细胞,包括多能干细胞和组织祖细胞,在分化成不同细胞类型的能力方面更加有限。
36.在本发明的方法中,基因工程化干细胞和/或祖细胞以过表达(即,以高于细胞正常水平的水平表达)treg谱系定型因子(例如,foxp3、helios、ikaros)和/或cd4
辅助因子t细胞谱系定型因子(例如,gata3和thpok)。这些因子促进了工程化的干细胞和/或祖细胞分化为treg。这些因子可能在整个或部分treg分化过程中组成性过表达;或者可以在treg分化过程的特定时期诱导地表达(例如,经由强力霉素诱导的tetr介导的基因表达)。
37.在一些实施方案中,定型因子由随机整合到干细胞或祖细胞基因组中的转基因编
码(例如,通过使用慢病毒载体、逆转录病毒载体或转座子)。
38.或者,定型因子由以位点特异性方式整合到干细胞或祖细胞基因组中的转基因编码。例如,将转基因整合在基因组安全港位点,或t细胞特异性基因的基因组基因座,如t细胞受体α链恒定区(即,t细胞受体α恒定区或trac)基因。在前一种方法中,转基因可以任选地置于t细胞特异性启动子或诱导启动子的转录控制之下。在后一种方法中,转基因可以在内源启动子和t细胞特异性基因的其他转录调控元件(例如,tcrα链启动子)的控制下表达。将转基因置于t细胞特异性启动子的控制下的优点是,转基因将仅在t细胞中表达,正如它所预期的那样,从而增强了工程化的细胞的临床安全性。
39.在一些实施方案中,本方法可以额外地包括进一步促进这种分化的组织培养步骤。
40.调节性t细胞维持免疫稳态并赋予免疫耐受。工程化的treg细胞可以是自体的或异体的,可用于基于细胞的疗法,以治疗需要诱导免疫耐受或恢复免疫稳态的患者,如接受器官移植或同种异体细胞疗法的患者以及患有自身免疫疾病的患者。目前的treg细胞将具有改善的治疗效果,因为它们可以是单克隆的,避免了过去treg疗法中由多克隆性引起的变异性。进一步,可以基于它们的抗原特异性来选择treg细胞。例如,可根据选择treg细胞以在需要treg的体内位点表达对抗原特异的t细胞受体(tcr)或编辑的嵌合抗原受体(car),以使tcr或car引导treg细胞到该位点(例如,炎症位点),从而增强细胞的效力。
41.i.编码cd4
treg定型因子的转基因
42.为了促进祖细胞或干细胞(如ipsc)分化为treg,可对细胞进行工程化以表达一种或多种蛋白质,这些蛋白质促进祖细胞或干细胞成为cd4
辅助t细胞并最终成为treg细胞的谱系定型。如本文所用,术语“调节t细胞”、“调节t淋巴细胞”和“treg”是指调节免疫系统、维持对自身抗原的耐受并且通常抑制或下调诱导和增殖t效应细胞的t细胞亚群。treg表型部分依赖于主转录因子叉头盒p3(foxp3)的表达,它调节免疫抑制功能所必需的基因网络的表达(参见,例如,fontenot et al.,nature immunology(2003)4(4):330-6)。treg通常由cd4
cd25
cd127
lo
foxp3
的表型来标志。在一些实施方案中,treg也是cd45ra
、cd62l
hi
、helios
和/或gitr
。在特定实施方案中,treg由cd4
cd25
cd127
lo
cd62l
或cd4
cd45ra
cd25
hi
cd127
lo
来标志。
43.在本发明的方法中,引入到干细胞或祖细胞的基因组以促进其分化为treg的转基因可以是但不限于编码以下项中一个或多个的那些:cd4、cd25、foxp3、cd4ra、cd62l、helios、gitr、ikaros、ctla4、gata3、tox、ets1、lef1、rora、tnfr2和thpok。编码这些蛋白质的cdna序列可在genbank和其他众所周知的基因数据库中获得。一种或多种这些蛋白质的表达在分化过程中有助于将干细胞或祖细胞定型为treg命运。在一些实施方案中,转基因编码treg谱系定型因子foxp3和/或cd4
辅助t细胞谱系定型因子thpok(he et al.,nature(2005)433(7028):826-33)。在一些实施方案中,转基因编码helios,其在treg亚群中表达(thornton et al.,eur j immunol.(2019)49(3):398-412)。
44.在一些实施方案中,工程化干细胞或祖细胞可以过表达增强造血干细胞(hsc)多能性的定型因子(参见sugimura et al.,nature(2017)545(7655):432-38)。这些因子包括但不限于hoxa9、erg、rora、sox4、lcor、hoxa5、runx1和myb。
45.在一些实施方案中,经由工程化的位点特异性转录抑制构建体(例如zfp-krab、
crispri等)、shrna或sirna,可以工程化干细胞或祖细胞以下调ezhi,以增强hsc多能性(参见vo et al.,nature(2018)553(7689):506-510)。
46.ii.转基因编码定型因子的整合
47.为了对干细胞或祖细胞进行基因工程化,将携带感兴趣的转基因的异源核苷酸序列引入细胞中。此处的术语“异源”是指把该序列插入基因组的位点中,其中该序列不是天然存在的。在一些实施方案中,将异源序列引入在treg细胞中具有特异性活性的基因组位点。此类位点的实例是编码t细胞受体链(例如,tcrα链、β链、γ链或δ链)、cd3链(例如,cd3 zeta、ε、δ或γ链)、foxp3、helios、ctla4、ikaros、tnfr2或cd4的基因。
48.例如,将异源序列引入基因组中的一个或两个trac等位基因。trac基因座的基因组结构在图1和2中阐释。trac基因位于tcrα链v和j基因的下游。trac含有三个外显子,它们被转录到tcrα链的恒定区。人trac基因的基因序列和外显子/内含子边界可在genbank id 28755或6955中找到。整合的靶位点可以是例如在内含子(例如,内含子1或2)内、在trac基因的最后外显子的下游区、在外显子(例如,外显子1、2或3)中或在内含子与其相邻外显子之间的连接处。
49.图1和2阐释了通过基因编辑将异源序列靶向人trac基因座的外显子2的两种不同方法。在这两种方法中,转基因编码含有一种或多种treg定型或诱导因子(例如foxp3)的多肽,由自切割肽(例如p2a、e2a、f2a、t2a)分离。在一些实施方案中,工程化foxp3转基因以将已知乙酰化的赖氨酸残基转化为精氨酸残基(例如,k31r、k263r、k268r),从而增强treg抑制活性(参见kwon et al.,j immunol.(2012)188(6):2712-21)。
50.在图1所示的方法中,工程化的细胞中tcrα链的表达被异源序列的插入破坏。在这种方法中,整合到基因组中的异源序列从5'到3'含有(i)自切割肽t2a的编码序列(或内部核糖体进入位点(ires)序列),(ii)定型因子的序列,和(iii)多聚腺苷酸化(polya)位点。一旦整合,工程化的trac基因座将在内源启动子下表达定型因子,其中t2a肽允许从第一个定型因子(即任何tcr可变结构域序列,以及任何由外显子1编码的恒定区序列和外显子2的5'到整合位点的部分)。由于trac基因被破坏,工程化的细胞中不能产生功能性tcrα链。由于在转基因中包含p2a编码序列,工程化的基因座可以将所有个体的treg诱导因子表达为单独的多肽。在这种方法下,可以进一步工程化干细胞或祖细胞以表达所需的抗原识别受体(例如,靶向感兴趣抗原的tcr或car)。
51.在图2所示的方法中,异源序列从5'到3'可以含有(i)trac外显子序列3'到整合位点(即整合位点下游的剩余外显子2序列,以及整个外显子3序列),(ii)t2a(或ires序列)的编码序列,(iii)一种或多种定型因子的编码序列,和(iv)polya位点。在异源序列中包含trac外显子序列和t2a将,这将允许产生完整的tcrα链。包含p2a将允许产生作为单独的多肽的定型因子。tcrα链、外源引入的定型因子均在内源tcrα链启动子的控制下表达。这种方法特别适用于工程化ipsc,该ipsc是从已经重排其tcrα和β链基因座的成熟treg重编程的(参见图7和下面的讨论)。从此类基因工程化的ipsc中分化出来的treg将保留祖先treg细胞的抗原特异性。进一步,tcrα链表达的保留可能会产生增强的t细胞和treg分化,因为tcr信号传导与胸腺中的t细胞和treg发育密切相关。
52.在替代的实施方案中,转基因可以整合到trac内含子中,而不是trac外显子中。例如,转基因整合在外显子2或外显子3上游的内含子中。在此类实施方案中,携带转基因的异
源序列从5'到3'可以含有剪接受体(sa)序列,该转基因编码一个或多个treg定型因子和polya位点。当需要表达重排的tcrα链基因时,异源序列可以从5'到3'含有(i)sa序列,(ii)异源序列整合位点下游的任何外显子,(iii)自切割肽或ires序列的编码序列,(iv)编码一种或多种定型因子的转基因,和(v)polya位点。一旦整合,sa将允许表达编码完整(即全长)tcrα链、自切割肽和定型因子的rna转录物。该rna转录本的翻译将产生两种(或更多)单独的多肽产物——完整的tcrα链和一个或多个定型因子。sa序列的实例是那些trac外显子和本领域已知的其他sa序列。
53.在一些实施方案中,将转基因整合到工程化细胞的基因组安全港中。基因组安全港位点包括但不限于aavs1基因座;rosa26基因座;clybl基因座;白蛋白、ccr5和cxcr4的基因座;以及将内源基因在工程化细胞中敲除的基因座(例如,t细胞受体α或β链基因座、hla基因座、ciita基因座或β2-微球蛋白基因座)。图3阐释了此类方法。在这个实例中,整合异源序列到人aavs1基因座中,例如,内含子1。编码转基因的定型因子的表达由强力霉素诱导启动子控制。强力霉素诱导启动子可以包括tet反应元件的5-mer重复。在将强力霉素引入组织培养物后,四环素控制的反式激活因子(rtta)的组成型表达诱导形式与tet反应元件结合并启动定型因子的转录。由引入的mrna产生的锌指核酸酶(zfn)在内含子1的特定位点(闪电)制造双链断裂。由质粒dna或线性化双链dna引入的供体序列从5'到3'含有同源区1、剪接受体(sa)剪接至aavs1外显子1、自切割肽2a的编码序列、嘌呤霉素抗性基因的编码序列、polya信号序列、5'基因组绝缘子序列、强力霉素诱导的定型因子盒、从cagg启动子驱动的rtta编码序列,然后是polya序列、3'基因组绝缘子序列和同源区2。基因组绝缘子序列确保其中的转基因不在分化过程中表观遗传沉默。同源区与zfn切割位点侧翼的基因组区同源。通过将嘌呤霉素引入培养物中,可以正向选择有成功靶向整合(ti)的细胞。treg诱导因子的诱导表达是有用的,因为某些因子在中胚层、造血或淋巴细胞发育过程中可能是有毒的,因此仅在t细胞发育期间打开这些因子以使分化偏向treg谱系是有利的。
54.在一些实施方案中,异源序列含有抗原结合受体,如嵌合抗原受体(car)的表达盒。图5和6阐释了此类实施方案的实例。在图5中,异源序列由质粒dna或线性化双链dna引入,并从5'到3'含有同源区1、car表达盒(在供体的反义方向)从其自身的启动子驱动并含有polya位点、5'loxp位点、剪接aavs1外显子1的剪接受体、自切割肽2a的编码序列、嘌呤霉素抗性基因的编码序列、自杀基因hsv-tk的编码序列、polya位点、5'基因组绝缘子序列、多西环素诱导定型因子表达盒、从cagg启动子驱动的rtta编码序列、4-羟基三苯氧胺(4-oht)诱导cre重组酶的编码序列,其中cre重组酶经由2a肽与rtta序列连接,然后是polya序列、3'基因组绝缘子序列、3'loxp位点和同源区2。基因组绝缘子序列确保其中的转基因不在分化过程中表观遗传沉默。同源区与zfn切割位点侧翼的基因组区同源。通过将嘌呤霉素引入组织培养物中,可以正向选择有成功靶向整合的细胞。组成型表达的4-oht诱导cre允许在向培养物中添加4-oht后切除loxp位点之间的整个盒。未经历重组酶介导切除的细胞仍将表达hsv-tk,因此可以通过将更昔洛韦(gcv)添加到组织培养物中来进行负向选择(消除)。gcv将导致任何表达hsv-tk的细胞死亡。该系统允许完全无疤痕地去除treg诱导盒,同时将car盒整合在一起,以允许在工程化的treg中进行靶向免疫抑制。
55.图6阐释了来自工程化的trac基因的car的表达。在本实例中,由质粒dna或线性化dsdna引入的异源序列从5'到3'含有同源区1、与car编码序列直接融合的2a编码序列,随后
是polya位点、5'loxp位点、5'基因组绝缘子序列、剪接至aavs1外显子1的剪接受体、2a编码序列、嘌呤霉素抗性基因的编码序列和自杀基因hsv-tk的2a肽连接编码序列,两者都脱离了它们自己的启动子,然后是polya信号序列、强力霉素诱导treg诱导因子表达盒、脱离cagg启动子的rtta编码序列、4-oht诱导cre重组酶的编码序列经由2a肽连接到rtta序列,然后是polya序列、3'基因组绝缘子序列、3'loxp位点和同源区2。基因组绝缘子序列确保其中的转基因不在分化过程中表观遗传沉默。同源区与zfn切割位点侧翼的基因组区同源。通过将嘌呤霉素引入组织培养物(可选地等待一周或更长时间以使未整合的供体附加体稀释出来),可以正向选择有成功靶向整合的细胞。组成型表达的4-oht诱导cre允许在向培养物中添加4-oht后切除loxp位点之间的整个盒。未经历重组酶介导切除的细胞仍会表达hsv-tk,因此可以通过将gcv添加到组织培养物中来消除。该系统允许完全无疤痕地去除treg诱导盒,同时使car盒远离整合的内源trac启动子,以允许在工程化的treg中进行靶向免疫抑制。
56.图11是描述整合crispr活化(crispra)或抑制(crispri)文库的基因组编辑方法的示意图,其包括分别与vph活化结构域或krab抑制结构域融合的死cas9(dcas9),驱动从强力霉素诱导启动子进入人aavs1基因的内含子1。在将强力霉素引入培养物后,四环素控制的反式激活因子(rtta)的组成型表达诱导形式与tet响应元件结合并启动整合的crispra或crispri构建体的转录。这些文库含有针对人类基因组中每个编码基因的grna,而每个细胞最多只能整合一个或两个dcas9-grna构建体(单等位或b等位基因靶向的整合)。由引入的mrna产生的zfn在内含子1的特定位点(闪电)发生双链断裂。由质粒dna或线性化dsdna引入的供体序列从5'到3'含有同源区1、剪接至aavs1外显子1的剪接受体、自切割肽2a的编码序列、嘌呤霉素抗性基因的编码序列、polya信号序列、5'基因组绝缘子序列、强力霉素诱导crispra或crispri构建体文库、从cagg启动子脱离的rtta编码序列,然后是polya序列、3'基因组绝缘子序列和同源区2。基因组绝缘子序列确保其中的转基因不在分化过程中表观遗传沉默。同源区与zfn切割位点侧翼的基因组区同源。通过将嘌呤霉素引入培养物中,可以正向选择有成功靶向整合的细胞。crispra或crispri构建体的可诱导表达是有用的,因为文库中靶向的某些基因的上调或下调在中胚层、造血或淋巴细胞发育期间可能是有毒的,因此仅在t细胞发育期间打开或关闭这些因子(在祖t细胞阶段后)向treg谱系倾斜分化是有利的,并且可能允许发现新的treg诱导因子和途径。
57.上述图仅用于阐释本发明的一些实施方案。例如,可以使用其他自切割肽代替图中所示的t2a和p2a肽。自切割肽是病毒衍生的肽,典型长度为18-22个氨基酸。自切割2a肽包括t2a、p2a、e2a和f2a。此外,2a肽的密码子多样化版本可用于在一个大的整合转基因盒上组合多个treg诱导基因。在一些实施方案中,使用ires代替自切割肽编码序列。内含子和外显子都可以作为靶。异源序列中可以包括额外的元件。例如,异源序列可以包括rna稳定元件,如土拨鼠肝炎病毒转录后调控元件(wpre)。
58.iii.基因编辑方法
59.可以使用任何用于将异源序列靶向整合到特定基因组位点的基因编辑方法。为了增强转基因位点特异性整合的精确度,携带异源序列的构建体可以在其一端或两端含有与靶基因组位点同源的同源区。在一些实施方案中,异源序列在5'和3'末端区都携带与t细胞特异性基因座或基因组安全港基因座中的靶基因组位点同源的序列。异源序列上的同源区
的长度可以是例如50-1,000个碱基对的长度。异源序列中的同源区可以但不必与靶基因组序列相同。例如,异源序列中的同源区可以与靶基因组序列(例如,由异源序列中的同源区取代的序列)有80%或更大百分比的同源或80%或更大百分比一致。在进一步的实施方案中,构建体在线性化时在一个末端包含同源区1,在其另一末端包含同源区2,其中同源区1和2分别与基因组区1和基因组区2同源,基因组区2在基因组中的整合位点的侧翼。
60.可以通过任何已知技术将携带异源序列的构建体引入靶细胞,如化学方法(例如磷酸钙转染和脂转染)、非化学方法(例如电穿孔和细胞挤压)、基于粒子的方法(例如、磁转染)和病毒转导(例如,通过使用病毒载体,如牛痘载体、腺病毒载体、慢病毒载体、腺相关病毒(aav)载体、逆转录病毒载体和杂合病毒载体)。在一些实施方案中,构建体是aav病毒载体并且通过重组aav病毒体将其引入靶人细胞,该重组aav病毒体的基因组包含该构建体,其包括在两末端具有aav反向末端重复(itr)序列以允许在生产系统(如昆虫细胞/杆状病毒生产系统或哺乳动物细胞生产系统)中产生aav病毒体。aav可以是任何血清型,例如aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav8.2、aav9或aavrh10,aav可以是任何假型,如aav2/8、aav2/5或aav2/6。
61.异源序列可以通过任何位点特异性基因敲入技术整合到trac基因组基因座中。此类技术包括但不限于同源重组、基于锌指核酸酶或切口酶(本文统称为“zfn”)、转录激活因子样效应核酸酶或切口酶(本文统称为“talen”)的基因编辑技术、成簇的规则间隔的短回文重复系统(crispr,如使用cas9或cpf1的系统)、大范围核酸酶、整合酶、重组酶和转座子。如以下工作实施例中所阐释的,对于位点特异性基因编辑,编辑核酸酶通常在靶向基因组序列中产生dna断裂(例如,单链或双链dna断裂),使得与靶基因组序列(例如,本文所述的构建体)同源的供体多核苷酸用作修复dna断裂的模板,导致将供体多核苷酸引入基因组位点。
62.基因编辑技术在本领域中是众所周知的。参见,例如,用于crispr基因编辑技术的美国专利8,697,359、8,771,945、8,795,965、8,865,406、8,871,445、8,889,356、8,895,308、8,906,616、8,932,814、8,945,839、8,993,233、8,999,641、9,790,490、10,000,772、10,113,167和10,113,167。参见,例如,用于zfn技术及其在编辑t细胞和干细胞中的应用的美国专利8,735,153、8,771,985、8,772,008、8,772,453、8,921,112、8,936,936、8,945,868、8,956,828、9,234,187、9,234,188、9,238,803、9,394,545、9,428,756、9,567,609、9,597,357、9,616,090、9,717,759、9,757,420、9,765,360、9,834,787、9,957,526、10,072,062、10,081,661、10,117,899、10,155,011和10,260,062。上述专利的公开内容通过引用整体并入本文。
63.在基因编辑技术中,可以通过改变复合物的dna结合特异性来定制基因编辑复合物以靶向特定的基因组位点。例如,在crispr技术中,可以设计引导rna序列以结合特定的基因组区;并且在zfn技术中,zfn的锌指蛋白结构域可以设计为具有对特定基因组区特异的锌指,使得zfn的核酸酶或切口酶结构域能够以位点特异性方式切割基因组dna。根据所需的基因组靶位点,可以相应地设计基因编辑复合物。
64.基因编辑复合物的组分可以通过众所周知的方法,如电穿孔、脂质体转染、显微注射、基因枪、病毒体、脂质体、脂质纳米颗粒、免疫脂质体、聚阳离子或脂质:核酸缀合物、裸dna或mrna以及人工病毒粒子,与转基因构建体同时或顺序递送到靶细胞中。使用例如
sonitron 2000系统(rich-mar)的声波穿孔也可用于核酸的递送。在特定实施方案中,基因编辑复合物的一种或多种组分,包括核酸酶或切口酶,作为mrna递送到待编辑的细胞中。
65.iv.treg的抗原特异性
66.在一些实施方案中,干细胞或祖细胞可以进一步工程化(例如,使用本文所述的基因编辑方法)以包括编码抗原识别受体,如tcr或car的转基因。或者,干细胞或祖细胞是从已经重排其tcrα/β(或δ/γ)基因座的成熟treg重编程的细胞,并且从此类干细胞或祖细胞重新分化的treg将保留他们祖先treg的抗原特异性。在任何情况下,treg可以针对它们对特定治疗目标的感兴趣抗原的特异性进行选择。
67.在一些实施方案中,感兴趣的抗原是多态性同种异体mhc分子,如由实体器官移植中的细胞或基于细胞的疗法(例如,骨髓移植、癌症car t疗法或基于细胞的再生疗法)的细胞来表达的。如此靶向的mhc分子包括但不限于hla-a、hla-b或hla-c;hla-dp、hla-dm、hla-doa、hla-dob、hla-dq或hla-dr。例如,感兴趣的抗原是i类分子hla-a2。hla-a2是移植中常见的错配组织相容性抗原。hla-a错配与移植后的不良结果有关。表达对mhc i类分子特异的car的工程化的treg是有利的,因为mhc i类分子在所有组织上广泛表达,因此无论移植的组织类型如何,treg都可用于器官移植。针对hla-a2的treg提供了额外的优势,即hla-a2在相当大比例的人群中表达,因此在许多供体器官上表达。有证据显示,hla-a2 car在treg细胞中的表达可以增强treg细胞在预防移植排斥中的效力(参见,例如,boardman,同上;macdonald et al.,j clin invest.(2016)126(4):1413-24;和dawson,同上)。
68.在一些实施方案中,感兴趣的抗原是自身抗原,即在身体特定组织的自身免疫炎症位点普遍或独特表达的内源抗原。对这种抗原特异的treg可以归巢于发炎的组织,并通过引起局部免疫抑制发挥组织特异性活性。自身抗原的实例是水通道蛋白水通道(例如,水通道蛋白4水通道)、副肿瘤抗原ma2、两性蛋白、电压门控钾通道、n-甲基-d-天冬氨酸受体(nmdar)、α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体(ampar)、甲状腺过氧化物酶、甲状腺球蛋白、抗n-甲基-d-天冬氨酸受体(nr1亚基)、rh血型抗原、桥粒芯蛋白1或3(dsgl/3)、bp180、bp230、乙酰胆碱烟碱型突触后受体、促甲状腺激素受体、血小板整合素、糖蛋白iib/iiia、钙帕他汀、瓜氨酸蛋白、α-β-晶状体蛋白、胃壁细胞内在因子、磷脂酶a2受体1(pla2r1)和含有7a的血小板反应蛋白1型结构域(thsd7a)。自身抗原的额外实例是多发性硬化相关抗原(例如,髓鞘碱性蛋白(mbp)、髓鞘相关糖蛋白(mag)、髓鞘少突胶质细胞糖蛋白(mog)、蛋白脂质蛋白(plp)、少突胶质细胞髓鞘寡聚蛋白(omgp)、髓鞘相关少突胶质细胞碱性蛋白(mobp)、少突胶质细胞特异性蛋白(osp/claudin 11)、少突胶质细胞特异性蛋白(osp)、髓鞘相关神经突生长抑制剂nogo a、糖蛋白po、外周髓鞘蛋白22(pmp22)、2'3'-环核苷酸3'-磷酸二酯酶(cnpase)及其片段);关节相关抗原(例如,瓜氨酸取代的环状和线性聚丝蛋白肽、ii型胶原蛋白肽、人软骨糖蛋白39肽、角蛋白、波形蛋白、纤维蛋白原和i、iii、iv和v型胶原蛋白肽);和眼相关抗原(例如,视网膜抑制蛋白、s-抑制蛋白、光感受器间类视黄醇结合蛋白、β-晶状体蛋白b1、视网膜蛋白、脉络膜蛋白及其片段)。在一些实施方案中,treg细胞靶向的自身抗原是il23-r(用于治疗例如克罗恩病、炎性肠病或类风湿性关节炎)、mog(用于治疗多发性硬化症)或mbp(用于治疗多发性硬化症)。在一些实施方案中,treg可以靶向其他感兴趣的抗原(例如,b细胞标志物cd19和cd20)。
69.在一些实施方案中,识别外来肽(例如,cmv、ebv和hsv)而不是同种抗原的treg可
用于同种异体过继细胞转移环境中,而没有通过识别同种抗原而不断被激活的风险,没有敲除tcr表达的需要。
70.v.用于基因组编辑的细胞
71.本公开的工程化的细胞是哺乳动物细胞,如人细胞、来自农场动物(例如,牛、猪或马)的细胞,和来自宠物(例如,猫或狗)的细胞。源细胞,即在其上进行基因组编辑的细胞,可以是全能干细胞(psc)。psc是能够在体内产生任何细胞类型的细胞,包括例如胚胎干细胞(esc)、通过体细胞核移植衍生的psc和诱导psc(ipsc)。分化ipsc和t细胞,参见,例如iriguchi and kaneko,cancer sci.(2019)110(1):16
–
22。如本文所用,术语“胚胎干细胞”是指从早期胚胎中获得的全能干细胞;在一些实施方案中,该术语是指从先前建立的胚胎干细胞系获得的esc,并且不包括通过最近破坏人胚胎获得的干细胞。
72.在其他实施方案中,用于基因组编辑的源细胞是多能细胞,如中胚层干细胞、间充质干细胞、造血干细胞(例如,从骨髓或脐带血中分离的那些)或造血祖细胞(例如,淋巴祖细胞)。多能细胞能够发育成不止一种细胞类型,但细胞类型潜能比全能细胞更受限制。多能细胞可以衍生自已建立的细胞系或可以从人骨髓或脐带分离。例如,造血干细胞(hsc)可在粒细胞集落刺激因子(g-csf)诱导的动员、普乐沙福诱导的动员或其组合之后从患者或健康供体中分离。为了从血液或骨髓中分离hsc,血液或骨髓中的细胞可能会被与不需要的细胞结合的抗体,如针对cd4和cd8(t细胞)、cd45(b细胞)、gr-1(粒细胞)和iad(分化的抗原呈递细胞)的抗体淘选(参见例如inaba,et al.(1992)j.exp.med.176:1693-1702)。然后可以通过cd34抗体正向选择hsc。
73.在一些实施方案中,待工程化的细胞是从成熟treg重编程的ipsc(takahashi et al.(2007)cell 131(5):861-72),如表达靶向非同种异体抗原的tcr的成熟treg。参见图7和下面的进一步讨论。
74.编辑的干细胞和/或祖细胞可以在移植到患者体内之前在体外分化成treg细胞。或者,干细胞和/或经编辑的祖细胞可以在移植到患者体内后诱导分化成treg细胞。
75.1.其它的基因组编辑
76.本发明的工程化细胞可以在上述基因组编辑之前或之后进行进一步的基因工程化,以使细胞更有效、更可用于更大的患者群体和/或更安全。基因工程化可以通过例如随机插入感兴趣的异源序列(例如,通过使用慢病毒载体、逆转录病毒载体或转座子)或靶向基因组整合(例如,通过使用由zfn、talen、crispr、位点特异性工程重组酶或大范围核酸酶介导的基因组编辑)。
77.例如,可以通过将car或tcr转基因位点特异性整合到细胞基因组中来对细胞进行工程化以表达一种或多种外源car或tcr。如上所述,外源car或tcr可以靶向感兴趣的抗原。
78.还可以编辑细胞以编码一种或多种治疗剂以促进treg的免疫抑制活性。治疗剂的实例包括细胞因子(例如,il-10)、趋化因子(例如,ccr7)、生长因子(例如,用于治疗多发性硬化的髓鞘再生因子)和信号传导因子(例如,双调蛋白)。
79.在其它的实施方案中,进一步工程化细胞以表达减少的细胞疗法的严重副作用和/或毒性的因子,如细胞因子释放综合征(crs)和/或神经毒性(例如,抗il-6scfv或分泌型il-12)(参见,例如,chmielewski et al.,immunol rev.(2014)257(1):83-90)。
80.在一些实施方案中,破坏工程化的细胞中的ezh1信号传导以增强它们的淋巴定型
(参见,例如,vo et al.,nature(2018)553(7689):506-10)。
81.在一些实施方案中,经编辑的细胞可以是患者的同种异体细胞。在此类情况下,可以进一步工程化细胞以减少宿主对这些细胞的排斥(移植排斥)和/或这些细胞对宿主的潜在攻击(移植物抗宿主疾病)。进一步工程化的同种异体细胞特别有用,因为它们可以用于多个患者而没有相容性问题。因此,同种异体细胞可以称为“通用”并且可以“现成”使用。使用“通用”细胞大大提高了效率并减少了采用细胞疗法的成本。
82.为了产生“通用”同种异体细胞,可以对细胞进行工程化,例如,使其具有以下项一种或多种的无效基因型:(i)t细胞受体(tcrα链或β链);(ii)多态性主要组织相容性复合物(mhc)i类或ii类分子(例如hla-a、hla-b或hla-c;hla-dp、hla-dm、hla-doa、hla-dob、hla-dq,或hla-dr;或β2-微球蛋白(b2m));(iii)与抗原加工相关的转运蛋白(例如,tap-1或tap-2);(iv)ii类mhc反式激活因子(ciita);(v)次要组织相容性抗原(miha;例如,ha-1/a2、ha-2、ha-3、ha-8、hb-1h或hb-1y);(vi)免疫检查点抑制剂,如pd-1和ctla-4;(vii)vim;和(vi)其任何组合。
83.同种异体工程化的细胞还可以表达不变的hla或cd47,以增加工程化的细胞(尤其是具有hla i类敲除或敲低的细胞)对宿主自然杀伤细胞和其他参与抗移植排斥的免疫细胞的抗性。例如,携带定型因子转基因的异源序列可以额外地包含不变hla(例如,hla-g、hla-e和hla-f)或cd47的编码序列。不变的hla或cd47编码序列可以通过自切割肽或ires序列的编码序列与异源序列中的初级转基因连接。
84.2.工程化的细胞中的安全开关
85.在细胞疗法中,移植的细胞可能需要在其基因组中含有“安全开关”,使得当患者不再需要细胞的存在时,可以停止细胞的增殖(参见,例如,hartmann et al.,embo mol med.(2017)9:1183-97)。例如,安全开关可以是自杀基因,其在向患者施用药物化合物时激活或失活,使得细胞进入凋亡。自杀基因可能编码在人中未发现的酶(例如,细菌或病毒酶),该酶在人细胞中将无害物质转化为有毒代谢物。
86.在一些实施方案中,自杀基因可以是来自单纯疱疹病毒(hsv)的胸苷激酶(tk)基因。tk可以将更昔洛韦、缬更昔洛韦、泛昔洛韦或其他类似的抗病毒药物代谢成有毒化合物,干扰dna复制并导致细胞凋亡。因此,宿主细胞中的hsv-tk基因可以通过向患者施用一种此类的抗病毒药物来开启以杀死细胞。
87.在其他实施方案中,自杀基因编码例如其他胸苷激酶、胞嘧啶脱氨酶(或尿嘧啶磷酸核糖基转移酶;将抗真菌药物5-氟胞嘧啶转化为5-氟尿嘧啶)、硝基还原酶(将cb1954(用于[5-(氮丙啶-1-基)-2,4-二硝基苯甲酰胺])转化为有毒化合物)、4-羟胺)和细胞色素p450(将异环磷酰胺转化为丙烯醛(氮芥))(rouanet et al.,int j mol sci.(2017)18(6):e1231)或诱导caspase-9(jones et al.,front pharmacol.(2014)5:254)。在额外的实施方案中,自杀基因可以编码细胞内抗体、端粒酶、另一种caspase或dna酶。参见例如zarogoulidis et al.,j genet syndr gene ther.(2013)doi:10.4172/2157-7412.1000139。
[0088]
安全开关也可以是“开启”或“加速器”开关、可以是编码小干扰rna、shrna或干扰对细胞存活至关重要的细胞蛋白表达的反义基因。
[0089]
安全开关可以利用任何合适的哺乳动物和其他必要的转录调控序列。可以使用本
文所述的基因编辑技术或本领域已知的其他技术,通过随机整合或位点特异性整合将安全开关引入细胞。可能需要将安全开关整合到基因组安全港中,使得工程化的细胞的遗传稳定性和临床安全性得到维持。本公开中使用的安全港的实例是aavs1基因座;rosa26基因座;clybl基因座;白蛋白、ccr5和cxcr4的基因座;以及在工程化的细胞中敲除内源基因的基因座(例如,t细胞受体α或β链基因座、hla基因座、ciita基因座或β2-微球蛋白基因座)。
[0090]
vi.体外重编程和分化细胞
[0091]
本公开的细胞可以使用本领域已知的方法从成熟的treg细胞重编程和/或在组织培养中分化成treg细胞。下面描述的方法仅仅是阐释性的而不是限制性的。
[0092]
1.将treg细胞重编程为ipsc
[0093]
在某些实施方案中,用于基因工程化的源细胞是从成人、青少年或胎儿treg细胞重编程的诱导全能干细胞(takahashi et al.,cell(2007)131(5):861-72)。在这些实施方案中,重编程的干细胞将保留其原始treg表型的表观遗传记忆(kim et al.,nature(2010)467(7313):285-90),因此可以重新分化回具有比其他干细胞,例如从不同细胞类型重编程的干细胞更高效率的treg。从treg重编程的干细胞也会保留v(d)j重排的tcr基因座,这可能会进一步增强干细胞的treg分化潜力,因为v(d)j重组是t细胞个体发育过程中的发育障碍(参见,例如,nishimura et al.,cell stem cell(2013)12(1):114-26)。
[0094]
用于重编程的treg细胞可以从多种来源分离,来源包括外周血单核细胞(pbmc)、骨髓、淋巴结组织、脐带血、胸腺组织或脾组织。例如,可以使用众所周知的技术,例如ficoll
tm
分离、红细胞裂解和单核细胞消耗后通过percoll
tm
梯度离心、逆流离心淘析、白细胞分离术和随后的基于细胞表面标志物的磁性或流式细胞仪分离,从受试者采集的血液单元中分离treg。
[0095]
从分离的白细胞中进一步富集treg细胞可以通过正向和/或负向选择以及针对独特表面标志物的抗体的组合使用如流式细胞术细胞分选和/或涉及缀合珠的磁免疫粘附等技术来完成。例如,为了通过阴性选择增强cd4
细胞,单克隆抗体混合物通常可以包括针对cd14、cd20、cd11b、cd16、hla-dr和cd8的抗体。为了富集或正向选择treg,可以使用针对cd4、cd25、cd45ra、cd62l、gitr和/或cd127的抗体。
[0096]
在示例性和非限制性方案中,treg细胞可以如下获得(参见dawson et al.,jci insight.(2019)4(6):e123672)。经由rosettesep(stemcell technologies,15062)从人供体中分离cd4
t细胞,并在使用moflo astrios(beckman coulter)或facsaria ii(bd biosciences)分选活cd4 cd25
hi cd127
lo
treg或cd4
cd127
lo
cd25 hi
cd45ra
treg之前富集cd25 细胞(miltenyi biotec,130-092-983)。可以在有1000u/ml il-2(proleukin)的immunocult-xf t细胞扩增培养基(stemcell technologies,10981)中用l细胞和抗cd3单克隆抗体(例如okt3,ubc ablab;100ng/ml)刺激分类的treg,如macdonald et al.,j clin invest.(2016)126(4):1413-24)中描述的。一天或多天后,treg细胞可被重编程(去分化)成干细胞,如下所述。对于表型分析,可以使用可固定活力染料(fvd,thermo fisher scientific,65-0865-14;biolegend,423102)对细胞进行染色,对于表面标志物,在固定和透化前使用ebioscience foxp3/转录因子染色缓冲液套装(thermo fisher scientific,00-5523-00)并染色细胞内蛋白质。在cytoflex(beckman coulter)上读取样品。
[0097]
然后可以使用重编程因子如oct3/4、sox2、klf4和c-myc(或l-myc)将treg重编程
为ipsc(参见,例如,nishino et al.,regen ther.(2018)9:71-8)。重编程因子可以经由非整合方法(例如仙台病毒、质粒、rna、小环、aav、idlv等)或整合方法(例如慢病毒、逆转录病毒和核酸酶介导的靶向整合)递送。
[0098]
图7阐释了将成熟treg重编程为ipsc的过程,ipsc随后被扩增,并以高效率重分化为treg细胞。这个过程从单treg细胞提供了扩大的、“恢复活力”的treg细胞池。
[0099]
2.干细胞向cd4
treg谱系的倾斜分化
[0100]
由于其基因组中存在编码定型因子的转基因,工程化的干细胞具有增加的treg分化潜能。图8阐释了其中ipsc分化成treg细胞的逐步分化过程:ipsc、中胚层干(祖)细胞、hsc、淋巴祖细胞、祖t细胞、未成熟单阳性(cd4
或cd8
)t细胞、双阳性t细胞(cd4
cd8
)、成熟的cd4
t细胞、最后是treg细胞。为了使这些干细胞分化为cd4
t细胞并最终成为treg细胞,可以采用组织培养技术。
[0101]
在一些实施方案中,干细胞在t细胞发育的后期阶段受到il-7ra(cd127)信号传导阻断以偏向分化为cd4
t细胞和treg谱系(参见singer et al.,nat rev immunol.(2008)8(10):788-801;图8和图9)。在其他实施方案中,ccr7信号传导在t细胞发育期间被阻断。与cd4
t细胞相比,ccr7在cd8
t细胞中被上调并促进t祖细胞对cd8
命运的定型(参见yin et al.,j immunol.(2007)179(11):7358-64)。在某些实施方案中,降低il-2浓度以向表达高水平高亲和力il-2受体(cd25)的treg提供增殖性生长优势(singer,同上;图8)。在某些实施方案中,与效应t细胞相比,优先促进treg增殖的活化珠用于激活和扩增treg(例如来自thermo fisher scientific的treg xpander珠)。
[0102]
图10阐释了采用的额外的组织培养技术。在其中阐释的一些实施方案中,工程化的干细胞与间充质基质细胞共培养(参见di ianni et al.,exp hematol.(2008)36(3):309-18)。此类基质细胞的实例包括促进淋巴定型的op9或op9-dll1基质细胞(参见hutton et al.,j leukocyte biology(2009)85(3):445-51;图10)。在其他实施方案中,胚胎中胚层祖细胞由全能干细胞形成并且经由在ms5-dll1/4细胞或epcam-cd56
基质细胞上的共培养在三维胚胎中胚层类器官中培养(图10)。然后将这些胚胎中胚层祖细胞分化成人造胸腺类器官,以更准确地复制胸腺发育过程(montel-hagen et al.,cell stem cell(2019)24(3):376-89.e8;seet et al.,nat methods(2017)14(5):521-30)。
[0103]
3.维持treg表型
[0104]
可塑性是几乎所有类型的免疫细胞所固有的特性。似乎treg细胞能够在炎症和环境条件下转变(“漂移”)为teff细胞(参见sadlon et al.,clin transl immunol.(2018)7(2):e1011)。为了在工程化的treg细胞中维持treg表型和/或增加转基因(例如,foxp3、helios和/或thpok)的表达,可以在含有雷帕霉素和/或高浓度的il-2的组织培养基中培养细胞(参见,例如,macdonald et al.,clin exp immunol.(2019)doi:10.1111/cei.13297)。在一些实施方案中,为了与teff相比优先扩增treg,可以在含有低剂量il-2的组织培养基中培养细胞(参见,例如,congxiu et al.,signal transduct targ ther.(2018)3(2):1-10)。
[0105]
vii.工程化的treg细胞的用途
[0106]
本公开的基因工程化treg细胞可用于细胞疗法以治疗需要诱导免疫耐受或恢复免疫稳态的患者(例如,人患者)。术语“治疗”(“treating”、“treatment”)是指减轻或消除
个细胞)。在某些实施方案中,组合物的单给药单位包含约106、约107、约108、约109或约10
10
或更多个细胞。可以每两天一次、每三天一次、每四天一次、每周一次、每两周一次、每三周一次、每月一次或以建立患者内有足够工程化的treg细胞所必要的另外频率。
[0114]
还提供了包含如本文所述的任何锌指核酸酶或其他核酸酶和多核苷酸的药物组合物。
[0115]
除非本文另有定义,否则与本公开相关的科学和技术术语应具有本领域普通技术人员通常理解的含义。下文描述了示例性方法和材料,尽管与本文描述的那些相似或等效的方法和材料也可以用于本公开的实践或检测中。在冲突的情况下,以本说明书,包括定义为准。一般而言,本文所述的心脏病学、医学、药物和药物化学以及细胞生物学所使用的命名法和技术是本领域众所周知和常用的。酶促反应和纯化技术根据制造商的说明书进行,如本领域通常完成的或如本文所述。进一步,除非上下文另有要求,否则单数术语应包括复数,复数术语应包括单数。在整个说明书和实施方案中,词语“具有(have)”和“包含(comprise)”或如“具有(has、having)”、“包含(comprises、comprising)”的变体将理解为意指包含所述完整物或完整物组,但不排除任何其他完整物或完整物组。本文提及的所有出版物和其他参考文献通过引用整体并入。尽管本文引用了许多文件,但该引用并不构成承认任何这些文件构成本领域公知常识的一部分。如本文所用,应用于一个或多个感兴趣值的术语“大约”或“约”是指与所述的参考值相似的值。在某些实施方案中,该术语是指在任一方向上落入10%、9%、8%、7%、6%、5%、4%、3%、2%、1%或更小的所述参考值的值范围(大于或小于),除非另有说明或从上下文中可以明显看出。
[0116]
为了更好地理解本发明,列出了以下实施例。这些实施例仅用于阐释目的,不应解释为以任何方式限制本发明的范围。
实施例
[0117]
实施例1:将转基因整合到ipsc的aavs1基因座
[0118]
本实施例描述了实验,其中整合绿色荧光蛋白表达盒到图3所示的aavs1基因座中。在第7天经由电穿孔将aavs1 zfn mrna和供体质粒递送到ipsc中。一周后(第0天),将嘌呤霉素(0.3μg/ml)添加到组织培养物中,开始对已进行靶向整合的细胞进行正向选择。在第15天加入多西环素,并以3种不同剂量(0.3、1和3μg/ml)维持在培养物中,以诱导dox诱导gfp表达盒的表达。对照细胞在培养物中没有添加强力霉素。细胞在多西环素存在下维持13天。在此期间,3μg/ml剂量的强力霉素产生最高水平的诱导gfp转基因表达(94%;图4)。当多西环素存在于培养物中时,这种高水平的表达是持续的。从第15-28天起,将细胞维持在嘌呤霉素和强力霉素中,并进一步进行正向选择(通过靶向整合将等位基因从~50%增加到~70%)。
[0119]
实施例2:ipsc向cd4
treg谱系的倾斜分化
[0120]
为了使ipsc分化为cd4
t细胞并最终成为treg细胞,干细胞通过使用靶向il-7受体α单元(il-7ra)的抗体阻断通过il-7受体的信号传导。在t细胞发育的后期,抗il-7ra抗体以增加的浓度添加到细胞培养基中。两次重复实验(实验1和实验2)均显示添加抗il-7ra抗体增加了cd4
单阳性细胞的百分比(右下象限),达到6.9%(实验1)或7.7%(实验2)。与未处理细胞的2.81%或4.78%相比,同时降低了cd8
单阳性细胞的百分比(左上象限)(图
9)。
[0121]
实施例3:从tipsc和cd34衍生的ipsc生成t细胞
[0122]
该实施例描述了从成熟t细胞(treg、cd4
和cd8
细胞)(本文称为“tipsc”)重编程的ipsc与从cd34
hspc重编程的ipcs获得分化t细胞的效率的对比研究。
[0123]
为了重编程t细胞和cd34
hspc,外周血单核细胞(pbmc)通过白细胞分离术从健康人供体中获得。t细胞亚群在流式细胞仪(sony sh800)上经由(miltenyi biotec)先前磁性抗体介导富集大量t细胞以获得幼稚cd4
cd25
高
cd127
低
cd45ra
treg,大量cd4
t细胞,和大量cd8
t细胞。使用基于仙台病毒的重编程试剂盒(thermo fisher scientific)对这些t细胞亚群进行重编程。cd34
细胞通过从pbmc中富集。
[0124]
在衍生自幼稚treg、cd4
t细胞、cd8
t细胞或cd34
干细胞的至少两个不同克隆中进行了至少两个实验。允许ipsc在56天的时间内分化为t细胞。
[0125]
图12中的数据展示了tipsc有效地分化为表达cd3和tcrαβ的细胞(图a和b)。tipsc系中两种t细胞标志物的共表达超过20%。相比之下,只有5%的从cd34衍生的ipsc系分化的细胞表达cd3和tcrαβ。p值如下:幼稚treg=0.03、cd4=0.48、cd8=0.02。
[0126]
当从活细胞/单细胞门控时,分化的ipsc产生了各种t细胞亚群(图12,图c)。t细胞亚群为cd3
tcrαβ
细胞,并包括cd4sp、cd8sp、双阳性(cd4
cd8
)和双阴性细胞。数据显示,与cd34衍生的ipsc相比,只有幼稚treg和cd8衍生的ipsc产生的双阴性细胞显著著减少。p值如下:幼稚treg=0.004、cd8=0.03。
[0127]
相比之下,不能从cd34衍生的ipsc产生表达cd3和tcrαβ的亚群(图12,图d)。数据显示,与cd34衍生的ipsc相比,cd4衍生的ipsc产生的cd8sp细胞(这些细胞也是cd3
tcrαβ
)显著更多,(p值=0.02)。
[0128]
实施例4:ipsc衍生的t细胞中的foxp3和抗hla-a2 car表达
[0129]
该实施例提供了关于使用图2所阐释方法的基因编辑研究的数据。在本研究中,编辑的ipsc trac基因座从5'到3'含有(i)trac外显子序列3'到整合位点(即整合位点下游的剩余外显子2序列,以及整个外显子3序列);(ii)t2a的编码序列;(iii)(a)foxp3/helios/car、(b)foxp3/car、(c)foxp3或(d)gfp的编码序列;(iv)polya位点。tcrα链和转基因均在内源tcrα链启动子的控制下表达。为清楚起见,所有转基因编码序列在相邻转基因之间含有框内的2a自切割肽编码序列,以允许多顺反子表达。
[0130]
分化编辑后的ipsc为cd34
造血干祖细胞(hspc),然后使用stemspan
tm
t细胞生成试剂盒(stemcell technologies)分化为dp t细胞(在ldcm上扩增2周,成熟1周)。dp t细胞通过用可溶性cd3/cd28/cd2激活剂刺激进一步分化。通过用荧光标记的hla-a2右旋体温育细胞来测定car表达。
[0131]
数据显示,在trac基因座编辑的ipsc衍生的t细胞中,转基因构建体引入的部分tcr编码序列能够维持tcrαβ表达,并且foxp3和car转基因也在这些细胞中过表达(图13a)。
[0132]
进一步评估了ipsc衍生的t细胞的细胞因子产生概况。在luminex flexmap仪器上分析细胞因子分泌(il-10、ifn-γ、tnf-α和il-2)之前,将细胞在200μl x-vivo
tm
培养基(lonza)中培养3天。将细胞因子浓度标准化为接种到培养物中的总活细胞。数据显示,在含有编辑过的foxp3或foxp3-2a-car转基因构建体的ipsc衍生的t细胞中,il-10的分泌增加,il-10是通过抑制效应t细胞的分化/活化,与调节性t细胞的抑制功能相关的免疫抑
制细胞因子(图13b)。尽管没有观察到tnf-α分泌的重大差异,但在含有foxp3和foxp3-2a-car转基因构建体的细胞中,il-2和ifn-γ分泌减少。il-2对于促进效应t细胞的存活和增殖很重要,而il-2的消耗是调节t细胞实现其抑制功能的一种机制。已显示由活化的t细胞产生的ifn-γ受到调节性t细胞的抑制。因此,这里的结果展示来自编辑到内源trac基因座中的foxp3转基因的foxp3过表达能够赋予编辑的t细胞treg样的表型。编辑后的细胞可以表达内源tcr和car(其中car是转基因构建体的一部分)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
