dna聚合酶和dna聚合酶衍生的3
′‑5′
外切核酸酶
技术领域
1.本发明涉及具有dna聚合酶和3
′‑5′
外切核酸酶活性的酶。具体而言,本发明涉及具有海洋来源的dna聚合酶ii活性和3
′‑5′
外切核酸酶活性的热不稳定酶(heat labile enzyme)。此外,本发明涉及主要发挥3
′‑5′
活性,即不存在聚合酶活性的dna聚合酶。本发明还涉及外切核酸酶活性,例如,在重组克隆过程中或在去除污染核酸分子的过程中降解双链dna的3
′‑5′
链而进行单链悬突(single stranded overhang)的用途。
背景技术:
2.合成生物学是一个快速发展的领域,被誉为应对未来生物经济和生物能源挑战的可能解决方案。合成生物学的最终愿景是创造细胞的新生物操作系统,能够可预见地执行有用的任务。合成生物学管道中的关键步骤之一是将dna片段组装成更大的通常会涉及多个组件的功能结构。
3.然而,当前的瓶颈是缺乏一种稳健的室温方法进行多dna组装而无需耗时的手动处理步骤。因此,非常需要一种能够绕过当前障碍的新dna组装方法。
4.基因组dna的复制是dna聚合酶的主要功能,它会催化单脱氧核糖核苷三磷酸(dntp)合成多脱氧核糖核苷酸。
5.在体外,dna聚合酶的特性用于dna合成,如用于各种dna扩增过程和dna分子的合成,读取所关注的dna链模板,产生与模板匹配的两条新dna链。
6.不同类型的聚合酶已被发现。例如,在大肠杆菌和其他原核细胞中,已知的dna聚合酶通常称为dna聚合酶i-v。各个组在复制保真度、热稳定性、延伸率以及校对活性(proof-reading activity)和效率方面各不相同。一些dna聚合酶相当简单,而另一些则更复杂,如由20个不同肽亚基组成的大肠杆菌聚合酶iii。
7.许多广泛使用的dna聚合酶在高温如最高达至少70℃下是稳定的,因此能够用于dna检测和分析方法,如聚合酶链式反应(pcr)或热循环dna测序。适用于此类过程的dna聚合酶通常称为热稳定dna聚合酶。
8.当用于体外dna复制过程时,除dntp外,还需要引物(初始寡核苷酸),携带能够用作链生长起点的3
′
末端羟基,因为dna聚合酶不能启动单核苷酸的从头合成。引物可以是带有游离3
′‑
oh基团的dna或rna的短片段或长片段,通过退火到模板的互补区域而为dna聚合酶提供双链结构。所选定的dna聚合酶沿模板工作,将引物沿5
′→3′
方向延伸。
9.由于dna链的极性,dna分子的两条链的复制是双向的,根据模板复制的方向,会产生两种不同的产物,一条“前导(leading)”链和一条“后随(lagging)”链。前导链合成为单条连续链,而所述后随链最初合成为小寡核苷酸,称为冈崎(okazaki)片段,然后连接形成连续链。在体内,小rna分子作为天然引物在前导链的合成中和特别是在后随链的合成中发挥作用。
10.众所周知,dna聚合酶iii连续合成前导链以及后随链上的冈崎片段,在合成片段之间留下间隙,然后由dna聚合酶i填充。
11.除了dna合成活性外,dna聚合酶还可能发挥其他酶活性,如3
′‑5′
外切核酸酶活性或链置换活性。在体内,一些dna聚合酶的3
′‑5′
外切核酸酶活性对于遗传稳定性,纠正dna聚合酶错误,例如,导致生成的dna分子中碱基对错配然后通过dna聚合酶的外切核酸酶功能进行校正的错误,是很重要的。已知dna聚合酶ii具有有效的3
′‑
5外切核酸酶活性,例如,纠正由dna聚合酶iii产生的错配错误,并且还被认为参与dna合成后损伤如,例如,由于uv照射的损伤的修复。
12.为了替换和纠正错配的碱基对,dna聚合酶的校对活性必须能够去除错误引入的dntp,因此所述核酸酶活性涉及dna分子磷酸骨架中的磷酸二酯键断裂。去除错配的dntp从而降解dna的能力在体外分子生物学中以各种方式被利用。
13.海洋来源的各种酶是已知的。例如,wo2017/162765公开了一种从冷杆菌属(psychrobacillus sp.)中分离出的海洋来源的热稳定dna聚合酶,在很宽的温度范围内,包括高于室温的温度,都有活性。
14.wo2016026574公开了一种源自冷水环境的热不稳定外切核酸酶,能够降解单链dna,并且如果暴露于低于65℃的温度时在15-20分钟内就可以失活。
15.本发明人已鉴定出一种源自粘放线菌(moritella viscosa)的热不稳定dna聚合酶ii,令人惊讶地发现其在不存在dntp的情况下具有非常强的3
′‑5′
外切核酸酶活性。本发明的酶是在来自受冬季溃疡病影响的养殖大西洋鲑鱼的粘放线菌菌株中鉴定出,这正如下文实验部分中进一步公开。
16.具体而言,据发现,本发明外切核酸酶能够结合双链dna分子并在3
′‑5′
方向降解其末端,导致受到本发明的dna聚合酶衍生的3
′‑
5外切核酸酶的dna分子的两端都产生5
′
悬突。
17.此外,还发现本发明的所鉴定的酶在室温下具有非常差的聚合酶活性。
18.所述已鉴定的dna聚合酶衍生的3
′‑5′
外切核酸酶的另一个优点是最佳活性的温度为约室温。此外,本发明的酶已经证实在高于25℃,如高于约30℃的温度下容易失活,导致所述外切核酸酶活性如果在约25℃的温度下使用时会在一段时间后停止。因此,例如,在用于制备5
′
悬突时,例如,在约5-30分钟内,可以形成合适长度的悬突。
[0019]3′‑
5外切核酸酶活性、聚合酶活性差和热不稳定性的组合会使该酶可用于分子克隆、多核苷酸去除和dna组装过程,如下所示。
[0020]
然而,本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶的一个优点是所述外切核酸酶活性在dntp存在下也是有活性的。因此,本发明的酶可用于,例如,制备5
′
悬突,而无需旨在去除dntp的预纯化过程。
[0021]
为了能够仅利用外切核酸酶活性,本发明人还合成了其中所述聚合酶活性被充分削弱或缺失的本发明的dna聚合酶修饰变体。
技术实现要素:
[0022]
根据第一方面,提供了分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段,其中所述dna外切核酸酶基本上没有聚合酶活性并且其中所述酶在高于25℃的温度下,如在高于约30℃的温度下不可逆地失活。
[0023]
根据第二方面,提供了分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片
段,其中所述dna聚合酶包含seq id no.1的氨基酸序列或包含与seq id no.1全长序列具有至少60%的序列同一性的氨基酸序列。
[0024]
根据第三方面,提供了分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段,所述dna聚合酶包含seq id no.2的氨基酸序列,或包含与seq id no.2全长序列具有至少60%序列同一性的氨基酸序列。
[0025]
根据第二和第三方面的所述分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段可以包含与具有seq id no.1或seq id no.2全长序列至少70%一致性,如与具有seq id no.1或seq id no.2全长序列至少80%序列同一性,如与具有seq id no.1或seq id no.2全长序列至少90%序列同一性的氨基酸序列。
[0026]
根据第四方面,提供了分离的dna聚合酶或其酶活性片段,其中所述聚合酶包含在对应于氨基酸位置v440-y447和位置g519-a523的至少一个氨基酸区域中包含至少一个突变的氨基酸序列。
[0027]
根据上述方面的一个实施方式,所述dna聚合酶在对应于d442、s445和/或d568的氨基酸位置上包含至少一个突变,其中所述至少一个突变是对以下的取代:
[0028]-在对应于d442的位置具有疏水侧链的氨基酸;
[0029]-在对应于s445的位置具有疏水侧链的氨基酸;和/或
[0030]-在对应于d568的位置具有疏水侧链的氨基酸。
[0031]
例如,根据上述方面的一个实施方式,所述dna聚合酶衍生的3
′‑5′
外切核酸酶包含的氨基酸序列中在对应于seq id no.1和seq id no.2中所示的氨基酸序列的d442和/或d568的氨基酸位置上具有至少一个突变。
[0032]
此外,根据上述方面的另一个实施方式,所述dna聚合酶衍生的3
′‑5′
外切核酸酶可以包含的氨基酸序列中在对应于seq id no.1和seq id no.2中所示的氨基酸序列的d442和/或d568的氨基酸位置上具有至少一个突变,并且其中所述至少一个突变是针对在对应于d442、s445和/或d568的位置上具有疏水侧链的氨基酸的取代。
[0033]
此外,根据上述方面的另一个实施方式,所述dna聚合酶衍生的3
′‑5′
外切核酸酶可以包含的氨基酸序列中在根据seq id no.1或seq id no.2编号的位置442上的氨基酸选自由glu、asp、ala、gly、val、leu和ile组成的组中。
[0034]
此外,根据上述方面的另一个实施方式,所述dna聚合酶衍生的3
′‑5′
外切核酸酶可以包含的氨基酸序列中在根据seq id no.1或seq id no.2编号的位置658上的氨基酸选自由glu、asp、ala、gly、val、leu和ile组成的组中。
[0035]
此外,根据上述方面的另一个实施方式,所述dna聚合酶衍生的3
′‑5′
外切核酸酶可以包含的氨基酸序列中在根据seq id no.1或seq id no.2编号的位置445上的氨基酸选自由ser、arg、lys和his组成的组中。
[0036]
此外,根据上述方面的另一个实施方式,所述dna聚合酶衍生的3
′‑5′
外切核酸酶可以包含的氨基酸序列中在位置442、445和568上的氨基酸选自由以下组成的组中:
[0037]
seq id no.1的氨基酸位置氨基酸442asp,glu,ala,gly,val,leu,ile445ser,arg,lys,his568asp,glu,ala,gly,val,leu,ile
[0038]
条件是位置442(d442)、445(s445)和568(d568)的氨基酸不同时是分别地asp、ser和asp。
[0039]
此外,根据上述方面的另一个实施方式,所述dna聚合酶衍生的3
′‑5′
外切核酸酶可以包含的氨基酸序列中在位置442和568上的氨基酸选自由以下组成的组中:
[0040]
seq id no.1的氨基酸位置氨基酸442asp,glu,ala445ser,arg568asp,glu,ala
[0041]
条件是位置442(d442)、445(s445)和568(d568)的氨基酸不同时是分别地asp、ser和asp。
[0042]
在根据上述任一方面的一个实施方式中,所述分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段是选自包含以下氨基酸序列的一组dna聚合酶的衍生的3
′‑5′
外切核酸酶的dna聚合酶衍生的3
′‑5′
外切核酸酶,其中
[0043]-位置442的氨基酸是ala;
[0044]-位置568的氨基酸是ala;
[0045]-位置442的氨基酸是glu;
[0046]-位置568位的氨基酸是glu;
[0047]-位置442和568的氨基酸是ala;和
[0048]-位置445的氨基酸是arg,其中根据seq id no.1的氨基酸序列进行编号。
[0049]
根据上述实施方式的dna聚合酶衍生的3
′‑5′
外切核酸酶是基本上没有聚合酶活性的dna聚合酶ii衍生的3
′‑5′
外切核酸酶,并且其中所述酶在高于25℃的温度下,例如,在高于约30℃的温度下,会不可逆地失活。
[0050]
根据第六方面,提供了分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段,所述dna聚合酶衍生的3
′‑5′
外切核酸酶分别包含选自由seq id no3、4、5、6、7和8组成的组中的氨基酸序列,或包含与具有seq id no.3、4、5、6、7和8全长序列至少60%,如至少70%,如至少75%,如至少80%,如至少85%,如至少90%,如至少95%,如至少97%,如至少98%或99%序列同一性的氨基酸序列,条件是
[0051]-seq id no.3中位置442的氨基酸是ala;
[0052]-seq id no.4中位置568的氨基酸是ala;
[0053]-seq id no.5中位置442的氨基酸是glu;
[0054]-seq id no.6中位置568的氨基酸是glu;
[0055]-seq id no.7中位置442和568的氨基酸是ala;和
[0056]-seq id no.8中位置445的氨基酸是arg。
[0057]
根据任何上述方面的另一个实施方式,所述分离的dna聚合酶衍生的3
′‑5′
外切核酸酶是dna聚合酶ii衍生的3
′‑5′
外切核酸酶。
[0058]
根据另一个实施方式,提供了根据本发明的分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段,其中所述酶在高于25℃的温度下,如在高于30℃的温度下会不可逆地失活。
[0059]
根据第七方面,提供了一种组合物,其包含根据前述权利要求中任一项所述的分
离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段和缓冲剂。
[0060]
根据第八方面,提供了编码根据本发明的分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段的核酸分子。
[0061]
根据第八方面的一个实施方式,提供了一种核酸分子,其中所述分子分别是选自由seq id no.1、seq id no.2、seq id no.3、seq id no.no.4、seq id no.5、seq id no.6和seq id no.7组成的组中的核酸序列,或包含与具有seq id no.3、seq id no.4、seq id no.5、seq id no.6和seq id no.7全长序列至少60%序列同一性的氨基酸序列。
[0062]
根据第八方面的另一个实施方式,提供了一种核酸分子,其中所述核酸分子包含seq id no.9或与seq id no.9全长序列具有至少80%序列同一性的序列。
[0063]
根据第九方面,提供了一种表达载体,其包含编码根据权利要求1-14中任一项所述的分离的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段的核酸分子和用于由所述核酸分子编码的蛋白质序列的转录和翻译的必需调控序列。
[0064]
根据一个实施方式,所述表达载体可以分别包含选自由seq id no.1、seq id no.2、seq id no.3、seq id no.4、seq id no.5、seq id no.id no.6和seq id no.7组成的组中的核酸序列,或包含与具有seq id no.3、seq id no.4、seq id no.5、seq id no.6和seq id no.7全长序列至少60%序列同一性的氨基酸序列。
[0065]
根据第十方面,提供了包含一种或多种根据本发明的表达载体或一种或多种根据本发明的核酸分子的宿主细胞。
[0066]
根据第十一方面,提供了一种制备本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段的方法,其包括以下步骤:
[0067]
a)在适合于表达所编码的dna聚合酶的条件下培养包含一种或多种根据权利要求19-20中任一项所述的重组表达载体或一种或多种根据权利要求16-18中任一项所述的核酸分子的宿主细胞;
[0068]
b)从所述宿主细胞或所述培养基或上清液中分离出或获得dna聚合酶衍生的3
′‑5′
外切核酸酶。
[0069]
根据第十二方面,本发明还涉及本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段在重组克隆过程中的用途,其中所述外切核酸酶或其酶活性片段会提供双链dna分子的单链dna悬突。
[0070]
根据第十三方面,提供了一种从样品中去除污染多核苷酸的方法,所述方法包括使所述样品与本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段接触。
[0071]
根据第十四方面,提供了一种用于删除一个或多个靶双链核酸分子的片段的方法,该方法包括使一个或多个双链核酸分子与本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段接触,其中所述外切核酸酶沿双链核酸分子的3
′‑5′
方向切割核苷酸而产生互补单链5
′
悬突。
[0072]
根据第十五方面,提供了组装两个或更多个双链(ds)dna分子的方法,所述方法包括以下步骤:
[0073]
(a)提供两个或更多个待组装的dsdna分子,其中dsdna分子的末端共享序列同一性区域;
[0074]
(b)将所提供的两个或更多个dna分子与根据本发明的热不稳定dna聚合酶衍生的3′‑5′
或其酶活性片段接触,由此在所提供的dsdna分子的两端产生单链悬突;
[0075]
(c)在所述dna分子藉此通过步骤(b)中产生的悬突部分退火的条件下培养(a)的dna分子;
[0076]
(d)可选地使步骤(c)中提供的所述退火的分子与dna聚合酶接触并允许dna聚合酶填充间隙,其中所述dna聚合酶具有降低的、受损的或失活的链置换活性。
[0077]
根据第十五方面的一个实施方式,步骤(a)-(d)在恒温下进行。
[0078]
根据第十五方面的另一个实施方式,步骤(a)-(d)在18-25℃范围内的温度下进行。
[0079]
根据第十五方面的另一个实施方式,将步骤(c)或(d)的所组装的dna分子进一步转移到合适的宿主细胞中进行增殖。
[0080]
根据第十五方面的又一个实施方式,步骤(b)在18-25℃范围内的温度下进行约5-15分钟。
[0081]
根据上述第十四和第十五方面的一个实施方式,消化dsdna分子或ds核酸分子,所产生的悬突的长度为10-40个核苷酸。
[0082]
根据第十六方面,提供了一种将至少一个靶双链核酸分子插入受体核酸分子中而提供重组双链核酸分子的方法,其包括以下步骤:
[0083]
(a)使本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段与靶双链核酸分子接触,其中所述外切核酸酶在所述靶链核酸分子末端的3
′‑5′
方向切割核苷酸而产生互补单链5
′
悬突;
[0084]
(b)使根据本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段与双链受体核酸分子接触,其中所述外切核酸酶在所述受体核酸分子的末端的3
′‑5′
方向上切割核苷酸而产生互补单链5
′
悬突;
[0085]
(c)提供反应混合物,其包含步骤(a)和(b)的产物,dna聚合酶,能够与(a)和(b)的核酸分子的一部分退火的寡核苷酸引物,和核苷酸;
[0086]
(d)在寡核苷酸引物藉此与步骤(a)和(b)的核酸分子退火的条件下培养所述反应混合物,并且由此所述dna聚合酶通过聚合一个或多个核苷酸而延伸所述寡核苷酸引物以产生重组双链分子。
[0087]
根据第十六方面的一个实施方式,该双链受体核酸分子是载体。根据第十六方面的又一个实施方式,步骤(a)和(b)在18-25℃范围内的温度下进行。根据第十六方面还有的另一个实施方式,步骤(d)的dna聚合酶是热不稳定dna聚合酶。
附图说明
[0088]
图1表示来自大肠杆菌的dna聚合酶ii模型(pdb代码:1q8i),它是来自粘放线菌(m.viscosa)的dna聚合酶ii的同源蛋白,并图示说明了氨基酸d442、s445和d568的位置,以及c-端和n-端。
[0089]
图2显示了本发明的酶的dna和氨基酸序列。
[0090]
图3显示了本发明的酶(mv pol ii)的聚合酶活性与来自大肠杆菌的klenow片段酶(kf)和嗜热性的嗜热脂肪芽孢杆菌(bst)聚合酶的聚合酶活性相比较。
[0091]
图4显示了本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶的外切核酸酶活性。
[0092]
图5显示了本发明的所述分离的酶(mv pol ii)在不同温度下的外切核酸酶活性。
具体实施方式
[0093]
正如上所述,本发明人已经鉴定出一种新的海洋来源的热不稳定dna聚合酶,其具有3
′‑5′
外切核酸酶活性和非常差的聚合酶活性,使得所述酶尤其适用于分子克隆过程。
[0094]
在整个本技术中,当提及“本发明的酶”或“dna聚合酶衍生的3
′‑5′
外切核酸酶”时,应该理解为是指本发明的酶,正如所附权利要求中所公开的并在下面的说明书中描述的。例如,包含seq id no 2的酶或其酶活性片段,或与seq id no.2相比在整个序列内具有约60%序列同一性的序列。所述术语还包括与具有seq id no.2的氨基酸序列的所述分离的酶相比被修饰,例如,通过定点诱变而被修饰的酶,其中所述修饰的酶保留了酶3
′‑5′
外切核酸酶活性,但与粘放线菌中鉴定出的所公开的野生型dna聚合酶相比,dna聚合酶活性受损、降低或缺乏。
[0095]
正如下文将进一步所示,外切核酸酶活性可以用于提供具有能够轻松插入载体中而扩增或表达靶dna分子的5
′‑
3悬突的靶dna分子。它也可以用于多核苷酸去除过程,例如,用于涉及其中过量或残留多核苷酸非所需的蛋白质或其他材料纯化的方法中。
[0096]
由于酶是热不稳定的而因此在高于25℃的温度下,例如,在高于约30℃的温度下很容易失活,该酶能够在室温下使用,并且也不需要费力或额外的失活步骤。
[0097]
本发明的酶可以用于室温下进行的各种过程中。术语“室温”是本领域公认的术语并且包括18-25℃范围内的温度。
[0098]
据发现该酶在去除受污染的dna或形成5
′
悬突所需的时间内是有活性的。即,一旦所述靶dsdna与本发明的酶接触,由于其也在室温下的热不稳定性,长度约10-40个核苷酸的悬突将在酶停止对靶dna分子的消化之前进行制备。例如,在室温下,据发现该酶会在约5-30min内形成具有长度约10-40个核苷酸的dsdna分子的5
′
悬突。如果在纯化过程中用于去除污染核酸,则所需时间也可能因待纯化样品的类型和污染核酸的量而异。
[0099]
除非本文专门定义,所有使用的技术和科学术语具有与遗传学、生物化学和分子生物学领域的技术人员通常理解的相同含义。
[0100]
所有与本文描述的方法和材料相似或等效的方法和材料都可以用于本发明的实践或测试,本文描述了合适的方法和材料。本文提及的所有出版物、专利申请、专利和其他参考文献通过引用以其全部内容结合于本文中。如有冲突,则以本说明书(包括定义)为准。
[0101]
当指出数字限制或范围时,则包括端点。此外,数字限制或范围内的所有值和子范围都被专门包括在内,就像明确写出一样。
[0102]
根据第一方面,提供了在高于约30℃的温度下不可逆失活的dna聚合酶或其酶活性片段。
[0103]
dna聚合酶的表述“酶活性片段”应该理解为是指其中聚合酶的活性被保持的dna聚合酶,即与具有如seq id no.1-8中描述的氨基酸序列的dna聚合酶相比具有相同或至少相似的活性,尽管与seq id no.1-8中描述的序列相比去除了一个或多个氨基酸。技术人员将会认可,例如,在氨基酸序列的c-端或n-端,可以去除一个或多个氨基酸,而不影响蛋白质的活性。
[0104]
根据另一方面,提供了dna聚合酶衍生的3
′‑5′
外切核酸酶,其中所述酶基本上没
有聚合酶活性,并且此外在高于25℃的温度下,例如,在高于约30℃的温度下会不可逆地失活。
[0105]
根据第二个方面,提供了一种dna聚合酶或其酶活性片段,包含seq id no.1或seq id no.2的氨基酸序列,或包含与具有seq id no.1全长序列至少60%序列同一性的氨基酸序列。
[0106]
本发明提供如上所述的具有3
′‑5′
外切核酸酶活性的dna聚合酶或其酶活性片段,其中所述酶基本上没有聚合酶活性。使用定点诱变,获得了聚合酶活性降低的野生型酶变体。
[0107]
在文献blasco etal.,the journal of biological chemistry,vol 268,no.32,pp 24106-24113中,使用定点诱变研究了对小(66kda)单亚基dna聚合酶活性的影响,表明特意(motive)dx2slyp形成了聚合酶活性的活性位点的部分。
[0108]
表述“基本上没有聚合酶活性”应该理解为表示与野生型dna聚合酶相比,本发明的酶的聚合酶活性的活性位点受损或缺失,所述野生型dna聚合酶具有根据seq id no.2的氨基酸序列。例如,本领域技术人员将会认可,具有降低的聚合酶活性的dna聚合酶与具有seq id no.3-7的氨基酸序列的dna聚合酶衍生的3
′‑5′
外切核酸酶的聚合酶活性相似,具有受损的聚合酶活性,即基本上没有聚合酶活性。本领域技术人员还将认可,聚合酶活性能够使用实时分子信标测定法(real time molecular beacon assay)进行测量,这如summerer,methods mol.biol.,2008,429,225-235中公开,或能够如下实施例4中所示的修饰形式进行测定。
[0109]
根据还有的另一方面,本发明提供了一种包含seq id no.1的氨基酸序列或具有与seq id no.1全长序列具有至少60%序列同一性的氨基酸序列的酶或其酶活性片段。
[0110]
根据还有的另一方面,本发明提供了一种包含seq id no.2的氨基酸序列或具有与seq id no.2全长序列具有至少60%序列同一性的氨基酸序列的酶或其酶活性片段。
[0111]
正如上所述,提供了一种dna聚合酶,包含根据seq id no.1或seq id no.2的氨基酸,并且相比于野生型序列(seq id no.2),在对应于氨基酸位置g447-l453和g519-a523的区域中包含至少一个突变。
[0112]
技术人员将会认可,氨基酸根据侧链的化学特性进行分组。氨基酸通常分为疏水性或亲水性的,和/或具有极性或非极性侧链。氨基酸替换为具有相同生化特征的另一种氨基酸,通常称为保守替换。技术人员将会认可,保守取代能够被引入蛋白质的氨基酸序列中,例如,根据本发明的酶中,而不会改变所述酶的活性。因此,预期此类修饰将构成生物学等效的产物。
[0113]
氨基酸的保守取代包括以下组内的氨基酸之间进行的取代:
[0114]
·
val、ile、leu、met(具有疏水侧链的氨基酸)
[0115]
·
phe、tyr、trp(具有疏水侧链的氨基酸)
[0116]
·
arg、his、lys(具有带正电侧链的氨基酸)
[0117]
·
ala、gly(具有小侧链的氨基酸)
[0118]
·
ser、thr(具有不带电侧链的氨基酸)
[0119]
·
asn、gln(具有不带电侧链的氨基酸)
[0120]
·
asp、glu(具有带负电侧链的氨基酸)
[0121]
一般而言,保守氨基酸取代是指不改变其中进行氨基酸取代的蛋白质的相对电荷或大小特征的氨基酸取代,而因此很少改变蛋白质的三维结构,这是为什么生物活性都没有显著改变的原因。
[0122]
因此,技术人员将会认可,具有根据seq id no.1或seq id no.2的氨基酸序列的酶,其中位置442的氨基酸选自由asp、glu、ala、gly、val、leu、ile组成的组中和/或位置445的氨基酸选自由ser、thr、arg、his、lys组成的组中,和/或其中位置568的氨基酸选自由asp、glu、ala、gly、val、leu、ile组成的组中,条件是位置442(d442)、445(s445)和568(d568)的氨基酸分别不是asp、ser和asp,将会具有与根据seq id no.3-8的酶相同的或大致相同的聚合酶活性和3
′‑5′
外切核酸酶活性。
[0123]
此外,技术人员将会理解的是,可以缺失、插入或添加一个或多个氨基酸,而不会改变本发明的酶的活性。
[0124]
因此,应当理解的是,本发明涵盖如所附权利要求中公开的dna聚合酶衍生的外切核酸酶,其中可以引入如上所述的这种修饰(氨基酸的取代、缺失、插入和添加),而基本上不会改变酶的活性,即关于聚合酶活性和外切核酸酶活性。
[0125]
根据还有的另一方面,本发明提供了一种dna-聚合酶或其酶活性片段,包含选自由seq id no.3、4、5、6、7和8组成的组中的氨基酸序列,或包含与分别具有seq id no.3、4、5、6、7和8全长序列至少60%序列同一性的氨基酸序列。
[0126]
根据另一个方面,提供了一种酶,其包含与选自由seqid no.1、seq id no.2、seq id no.3、seq id no.4、seq id no.5、seq id no.6、7和seq id no.8组成的组中的氨基酸序列全长序列内至少60%序列同一性,如至少70%,如至少75%,如至少80%,如至少85%,如至少90%,如至少95%,如至少97%,如至少98%或99%序列同一性的氨基酸序列。
[0127]
此外,本发明还提供一种编码根据本发明的酶或其酶活性片段的核酸分子,以及与其基本上同源的核酸分子。
[0128]
根据一个方面,提供了一种编码选自由seq id no.1、seq id no.2、seq id no.3、seq id no.4、seq id no.5、seq id no.6和seq id no.7组成的组中的氨基酸序列或包含与具有seq id no.3、seq id no.4、seq id no.5、seq id no.6、seq id no.7和seq id no.8全长序列至少60%序列同一性的氨基酸序列的核酸分子。
[0129]
根据还有的另一方面,提供了一种核酸分子,其包含如seq id no.9所示的序列或与seq id no.9全长序列具有至少80%序列同一性,如与seq id no.9全长序列具有至少85%,如至少90%,如至少95%,如至少97%,如至少98%,如至少99%序列同一性的核酸分子。
[0130]
因此,技术人员将会认可,具有根据seq id no.1或seq id no.2的氨基酸序列的dna聚合酶,其中位置442的氨基酸选自由glu、asp、ala、gly、val、leu和ile组成的组中和/或位置445的氨基酸选自由ser、arg、lys和his组成的组中,和/或其中位置568的氨基酸选自由glu、asp、ala、gly、val、leu和ile组成的组中,可以具有与根据seq id no.3-8的dna聚合酶相同或大致相同的3
′‑
5外切核酸酶活性和降低或失活的聚合酶活性。
[0131]
此外,技术人员将会理解的是,可以缺失、插入或添加一个或多个氨基酸而不改变本发明的酶的活性。
[0132]
因此,应当理解的是,本发明涵盖如所附权利要求中公开的dna聚合酶,其中可以
引入如上所述的这种修饰(氨基酸的取代、缺失、插入和添加)而基本上不改变酶的活性,即根据本发明关于3
′‑
5外切核酸酶活性。
[0133]
正如本文所用,就蛋白质和核酸分子或其片段而言,当提及“序列同一性”时,与第二序列具有至少x%一致性的序列是指x%代表第一序列中相对于第二氨基酸序列的总长度,当两个序列通过全局比对进行最佳比对时,它们与第二序列的匹配氨基酸相同的氨基酸数量。当x最大时,两个序列都最佳比对。一致性百分比的比对和确定可以手动或自动进行。每当在本文中提及序列同一性时,应该理解的是,分别与seq id no.1-seq id no.9中所示的整序列进行比较。
[0134]
技术人员将会认可,为了确定百分比氨基酸序列同一性的目的的比对能够以本领域技术范围内的各种方式,例如,使用诸如clustalomega(sievers f,higgins dg(2018)protein sci 27:135-145)、protein blast(美国国家生物技术信息中心(national center for biotechnology information)(ncbi))的公开可获得的计算机软件或诸如megalign(dnastar)软件的市售软件进行实现。本领域技术人员能够确定用于测量比对的合适参数,包括在被比较的序列的全长内实现最大比对所需的任何算法。ncbi blast是另一个用于确定氨基酸序列同一性的软件实例(macwilliam et al.,nucleic acids res.2013jul;41(web server issue):w597
–
w600)。
[0135]
技术人员将会认可,在不改变氨基酸序列的核酸分子中能够引入修饰,例如,核苷酸的取代导致受取代影响的三联体仍然编码相同的氨基酸。例如,氨基酸异亮氨酸由三联体(dna密码子)att、atc和ata编码。随后,异亮氨酸三联体att中第三个核苷酸从t替代为c或a,将不会改变所得氨基酸序列。通过技术人员熟知的技术(例如,定点诱变)可以引入此类核苷酸修饰,以使核酸序列适应宿主细胞优选使用的密码子,从而增强酶的表达。
[0136]
此外,编码促进分离和纯化的多肽的核酸分子能够添加到本发明的核苷酸序列中而不影响所得酶的活性。
[0137]
此外,用于使宿主细胞分泌所需的酶而提供的编码信号肽的核酸分子也可以与本发明的核酸序列连接。
[0138]
本发明还包括包含本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶或其酶活性片段的组合物。该组合物还可以包含缓冲剂。本领域技术人员将会认可,用于包含本发明酶的组合物中的缓冲剂可以根据选择的酶和其中使用该酶的方法而变化和优化。本发明的酶的3
′‑5′
外切核酸酶活性保持于本领域技术人员熟知的分子克隆、dna组装和多核苷酸消化方法中常用的条件,即,例如,关于盐的类型和浓度、ph条件等内。
[0139]
例如,可以使用众所周知的缓冲剂,如tris缓冲剂,例如,具有ph高于约8.0的tris缓冲剂,例如,8.0-9.0范围内的ph的tris缓冲剂。根据一方面,组合物的ph处于8.5-9.0之内。
[0140]
此外,技术人员将会认可,盐的类型及其浓度可以变化。根据一方面,所述组合物包含一种或多种选自由nacl和kcl组成的组中的盐。根据本发明的另一方面,组合物包含nacl和kcl。根据还有的另一方面,该组合物包含至多约25mm的nacl和kcl。
[0141]
本发明的dna聚合酶的制备
[0142]
本发明的酶及其酶活性片段或对其编码的核酸分子从其自然环境中进行纯化或分离,或它们通过技术人员熟知的克隆操作和重组dna操作而产生。
[0143]
编码根据本发明的dna聚合酶衍生的3
′‑5′
外切核酸酶或编码其酶活性片段的核酸分子可以通过技术人员或商业供应商如,例如,genscript、thermo fisher scientific等众所周知的方法进行合成。
[0144]
本领域技术人员非常了解并熟悉用于表达所分离或纯化的核酸分子的各种可用生物技术,以通过使用常用基因工程技术和重组dna表达系统在各种宿主细胞系统中异源表达而制备重组蛋白,参见,例如,“recombinant gene expressionprotocols,in methods in molecular biology,1997,ed.rocky s tuan,human press(issn 1064-3745)或sambrook et al.,molecular cloning:alaboratory manual(third edition),2001,cshl press,(isbn 978-087969577-4)。例如,编码根据本发明的酶或编码其酶活性片段的核酸分子可以插入包含特别适合于指导所需蛋白质编码核酸序列在合适宿主细胞中表达的所有必要的转录和翻译调节序列的合适表达载体中。合适的表达载体是,例如,质粒、粘粒、病毒或人造酵母染色体(yac)。
[0145]
例如,将要表达和用于制备根据本发明的dna聚合酶的dna分子可以插入到用于扩增所关注的序列或用于表达本发明的dna聚合酶编码序列的载体中。快速克隆(fastcloning)是用于此目的的适用方法的一个实例。
[0146]
根据本发明的一个方面,提供了一种载体,如表达载体,其包含编码根据本发明的酶或其酶活性片段的核酸分子。
[0147]
根据一个进一步的方面,提供了一种载体,如表达载体,其包含编码选自由seq id no.1、seq id no.2、seq id no.3、seq id no.4、seq id no.5、seq id no.6、seq id no.7和seq id no.8组成的组中的氨基酸序列或具有与全长序列至少约60%序列同一性,例如,与seq id no.1、seq id no.2、seq id no.3、seq id no.4、seq id no.5、seq id no.6、7和seq id no.8全长序列至少70%、75%、80%、85%、90%、95%、96%、97%、98%或99%序列同一性的氨基酸序列的核酸分子。
[0148]
根据一个进一步的方面,提供了包含seq id no.9或与seq id no.9全长序列具有80%序列同一性,如与seq id no.9全长序列具有至少85%、90%、95%、96%、97%、98%或99%序列同一性的序列的载体。
[0149]
本领域技术人员将会认可,使用包含编码根据本发明的dna聚合酶的核酸分子的表达载体可以制备根据本发明的dna聚合酶,其中所述分子可操作地连接至适用于所关注的宿主细胞的启动子。
[0150]
本领域技术人员还将认可,正如本文所用的“启动子”是指控制和启动特定基因转录的dna编码序列的dna上游区域(5
′
)。启动子会控制rna聚合酶和其他蛋白质的识别和结合以启动转录。“可操作地连接”是指启动子和第二序列之间的功能连接,其中启动子序列启动和介导对应于第二序列的dna序列的转录。通常而言,可操作地连接是指被连接的核酸序列是连续的。例如,适用于细菌宿主细胞中表达重组蛋白的载体可以包含适用于细菌表达系统的启动子,如t7启动子。
[0151]
根据本发明的载体可以使用技术人员熟知的标准质粒分离技术,如例如,使用qiagen
tm
的qiaprep
tm spin miniprep试剂盒或qiagen
tm plasmid plus maxi试剂盒进行分离。
[0152]
包括编码本发明的酶或其酶活性片段的核酸分子的表达载体可以引入合适的宿
biology,2012,vol.852,pp 51-59,公开了用于生产重组dna的slic方案,其中靶dsdna使用t4dna聚合酶的外切核酸酶活性(在不存在dntp的情况下)插入所选择的载体中而产生要进行组合的dsdna的单链悬突,然后对所退火的产物进行退火和连接酶处理。
[0165]
基于同源重组的其他方法出现在,例如,ep1929012中,其公开了一种同源重组方法,其中dsdna分子在用产生悬突的3
′‑5′
外切核酸酶处理后进行组装,然后退火,使所退火的产物与dna聚合酶接触,并最后使用连接酶密封所述再切割切口,并且其中该过程在拥挤剂(crowding agent)(例如peg)的存在下进行。
[0166]
本发明的dna聚合酶也可以用于其他类似的方法,如ep1915446a1和ep2255013中公开的方法。
[0167]
多种dna组装系统也可商购获得,如例如,由thermofisher scientific提供的组装产品和由newengland biolabs inc.提供的neb gibsonhifidna assembly和golden gate assembly。
[0168]
为了以所需的顺序组装多个dna分子,要组装的末端应该共享序列同一性,以确保对由外切核酸酶消化步骤产生的所关注的相应悬突发生退火(杂合)。悬突的长度优选足以与序列同一性共享区域的互补悬突进行特异性杂合的长度,从而允许单链悬突进行杂合。作为退火多个dsdna分子的原理的说明,请参考slic:a method for sequence and ligationindependent cloning by li and elledge,2012,gene synthesis,pp 51-59中的第54页的图2。
[0169]
根据本发明,通过使待组装的dsdna分子与dna聚合酶衍生的3
′‑5′
外切核酸酶接触而制备的悬突的长度为10-40个核苷酸。
[0170]
根据一个方面,提供了一种方法,所述方法包括以下步骤:
[0171]
(a)提供两个或多个待组装的dsdna分子,其中dsdna分子的末端共享序列同一性区域;并且其中,对于要连接的每对dsdna分子,第一dna分子的远端区域和第二dna分子的近端区域共享包含10-40个核苷酸的序列同一性区域;
[0172]
(b)将所提供的两个或更多个dna分子与根据本发明的热不稳定dna聚合酶衍生的3
′‑5′
外切核酸酶接触,由此在所提供的dsdna分子的两端产生单链悬突;
[0173]
(c)在所述dna分子藉此通过步骤(b)中产生的悬突进行退火的条件下培养(a)的dna分子;
[0174]
(d)可选地使步骤(c)中提供的所退火的分子与dna聚合酶接触并允许所述dna聚合酶填充间隙,其中所述dna聚合酶具有降低的、受损的或失活的链置换活性,并发挥校对活性。
[0175]
本发明的酶是特别适合的,因为在室温下能够消化所关注的dsdna分子以提供单链悬突。此外,由于本发明的酶是热不稳定的并且在高于25℃的温度下失活,因此不需要费力的失活步骤。
[0176]
因此,本发明提供了用于组装两个或更多个双链(ds)dna分子的方法,如所附权利要求中所公开的。
[0177]
根据一个实施方式,所述在本发明的多dna组装方法中要组装的至少两种或更多种dsdna分子之一是载体。载体和待组装的dsdna分子可以与本发明的dna衍生的3
′‑5′
外切
核酸酶单独或一起在一个步骤中接触。
[0178]
本发明的酶的外切核酸酶活性也可以用于去除多核苷酸的方法,例如,用于纯化方法中。例如,该酶可以用于纯化从天然来源分离或在各种宿主系统中表达的多肽或蛋白质,其中多核苷酸代表杂质或污染物,并且其中包含所需蛋白质的溶液与本发明的酶一起培养,会提供所讨论的所需蛋白质的非多核苷酸纯化溶液。此外,使用本发明的酶可以纯化包含一种或多种所关注的受多核苷酸污染的试剂的其他溶液。当用于从包含蛋白质或其他试剂的溶液中去除多核苷酸时,本发明的一个优点是该酶可以在室温下使用,并且还不需要失活步骤,因此避免,例如,添加失活添加剂,如金属离子或螯合剂,它们可能对所讨论的蛋白质或试剂具有不良影响或其进一步的用途。
[0179]
实施例
[0180]
实施例1dna聚合酶ii衍生的3
′‑5′
外切核酸酶的克隆
[0181]
lunder等报道了(disease of aquatic organisms,vol.23,no.23,pp.39-49,1995)采用从受冬季溃疡病感染的养殖大西洋鲑鱼中分离出的弧菌(vibrio)样细菌进行实验。后来,据发现,所分离出的细菌是一种精神营养的海洋粘放线菌。
[0182]
提供编码本发明的酶的酶的基因是在使用所述粘放线菌的基因组dna(基因库:ln554852.1)的聚合酶链式反应中使用以下引物而获得。
[0183]
使用技术(thermo fisher)将已鉴定出的编码来自粘放线菌的dna聚合酶i的基因克隆到phmgwa载体中。聚合酶链式反应的起始材料是粘放线菌的基因组dna。使用quikchange ii定点诱变试剂盒(agilent technologies)将各种突变引入,并通过测序分析进行证实。
[0184]
正向引物
[0185]5′‑
caccttgtctgctacatatctgggt-3
′
(seq id no.10)
[0186]
反向引物
[0187]5′‑
ttaaaataatcccatttgttgatcggttatca-3
′
(seq id no.11)。
[0188]
为了提供修饰的酶,其中所鉴定的酶与野生型酶相比的聚合酶活性发生了降低、受损或失活,通过quikchange ii定点诱变试剂盒(agilent technologies)在所鉴定的克隆基因中引入了各种突变,并通过测序分析进行证实。
[0189]
实施例2:重组酶(mv pol ii及其突变体)的制备
[0190]
为了在宿主细胞中表达本发明的酶,将编码本发明dna聚合酶的基因序列(seq id no.2,针对所讨论的宿主细胞优化的密码子)被引入到gateway destination vector phmgwa中。
[0191]
根据本发明的酶的重组蛋白生产在rosetta 2(de3)细胞中进行。细胞在terrific broth培养基/氨苄青霉素(100μg/ml)中生长,并通过添加0.1mm iptg而在od
600nm 1.0下诱导基因表达。蛋白质生产在15℃下进行6-8小时。
[0192]
对于蛋白质纯化,将1-1培养物的颗粒再悬浮于50mm hepes ph 7.5(25℃)、500mm nacl、5%甘油、0.15mg/ml溶菌酶、1片蛋白酶抑制剂片剂(complete
tm
,mini,edta-free protease inhibitor cocktail,roche)并在冰上培养30min。
[0193]
使用的vcx 750通过超声处理进行细胞破碎(脉冲1.0/1.0,15分钟,幅度
25%)。在第一步中,在离心(48384g,45分钟,4℃)和过滤(φ0.45μm)后获得的his
6-标记蛋白的可溶性部分通过固定ni
2 -亲和色谱进行纯化。在用50mm hepes(25℃)、500mm nacl、35mm咪唑、5%甘油进行洗涤步骤后,通过使用脱盐柱以250mm的咪唑浓度洗脱蛋白质并进一步转移到50mm hepes(25℃)、500mm nacl、10mm mgcl2、5%甘油中。第二步是通过tev蛋白酶在50mm tris ph 8.0、0.5mm edta和1mm dtt中4℃下过夜处理切下标记。为了从his
6-标记和his
6-标记的tev蛋白酶中分离出蛋白质,在第三步中通过施加50mm hepes(25℃)、500mm nacl、5%甘油进行第二ni
2
亲和色谱。最终的蛋白质溶液进行浓缩并与50%甘油一起储存于-20℃下用于活性分析测试。
[0194]
实施例3:本发明的酶的聚合酶活性的测定
[0195]
为了测量本发明的酶的聚合酶活性并将所述新型酶与已知的dna聚合酶进行比较,使用了基于分子信标探针(改自summerer,methods mol.biol.,2008,429,225-235)的测试法。分子信标模板由23链节环组成,该环由富gc 8链节茎区(序列以斜体表示)和43链节延伸进行连接。由于环形成,荧光团dabcyl和fam非常接近并因此被淬灭。通过与分子信标模板退火的引物的dna聚合酶i延伸,茎被打开,并两个荧光团距离的增加通过fam荧光的恢复(激发485nm,发射518nm)进行测量。
[0196]
分子信标模板
[0197]5′‑
ggcccgt
dabcyl
aggaggaaaggacatcttctagcat
fam
acgggccgtcaagttcatggccagtcaagtcgtcagaaatttcgcaccac-3
′
(seq id.no.12)
[0198]
引物
[0199]5′‑
gtggtgcgaaatttctgac-3
′
(seq id.no.13)
[0200]
分子信标底物是通过将20μl 10μm分子信标模板和15μm引物在10mm tris-hcl ph 8.0、100mm nacl中95℃下培养5分钟而产生。然后让反应在室温下冷却2小时。底物溶液在-20℃下储存,而最终浓度为10μm。
[0201]
50微升反应由200nm底物和200μm dntp(等摩尔量的datp、dgtp、dctp和dttp)组成。该反应还包含5mm mgcl2在50mm tris-hcl ph 8.5、100mm kcl、1mm dtt、0.2mg/ml bsa和2%甘油中的溶液。活性测试在25℃下于黑色96孔荧光分析测试板中进行。通过添加蛋白质溶液,即mv pol ii及其变体引发反应。通过在485nm处激发和在518nm处发射而测量fam荧光的增加,作为适当时间间隔内的相对荧光单位。该测量在gemini microplate reader(molecular devices)中完成。结果如图3所示,并表明本发明的酶具有低dna聚合酶活性。
[0202]
实施例5:本发明的酶的3
′‑5′
外切核酸酶活性
[0203]
通过在10mm tris-hcl ph 8.0,100mm nacl中在75℃下将40μl 0.5μm模板dna与0.5μm fam标记的反向互补链(seq id no,14和seq id no.15)培养5分钟,产生用于外切核酸酶测试的平端dsdna底物。然后让反应在室温下冷却2小时。底物溶液在-20℃下储存,最终浓度为0.5μm。
[0204]
10微升反应含有25nm底物、5mm mgcl2在50mm tris-hcl ph 8.0、25mm nacl、1mm dtt、0.2mg/ml bsa和2%甘油中的溶液。通过添加0.02μg/μl蛋白质(即,mv pol ii及其变体)引发反应。作为阴性对照,使用蛋白质稀释缓冲剂代替蛋白质溶液。通过加入2.5μl变性凝胶上样缓冲剂(95%甲酰胺、10mm edta、0.1%二甲苯氰(xylene cyanol))并在95℃下培
养5分钟而终止反应。对于变性聚丙烯酰胺凝胶电泳(12%聚丙烯酰胺/7m脲),将6μl样品体积上样到凝胶上。凝胶电泳在0.5
×
tbe缓冲剂(44.5mm tris、44.5mm硼酸、1mm edta)中以50w进行1小时15分钟,并随后用pharosfx plus imager(bio-rad)扫描凝胶。
[0205]5′‑
[fam]tatccaccaatactaccctacgatactttgtccactcaat-3
′
(seq id.no.14)
[0206]3′‑
ataggtggttatgatgggatgctatgaaacaggtgagtta-5
′
(seq id.no.15)
[0207]
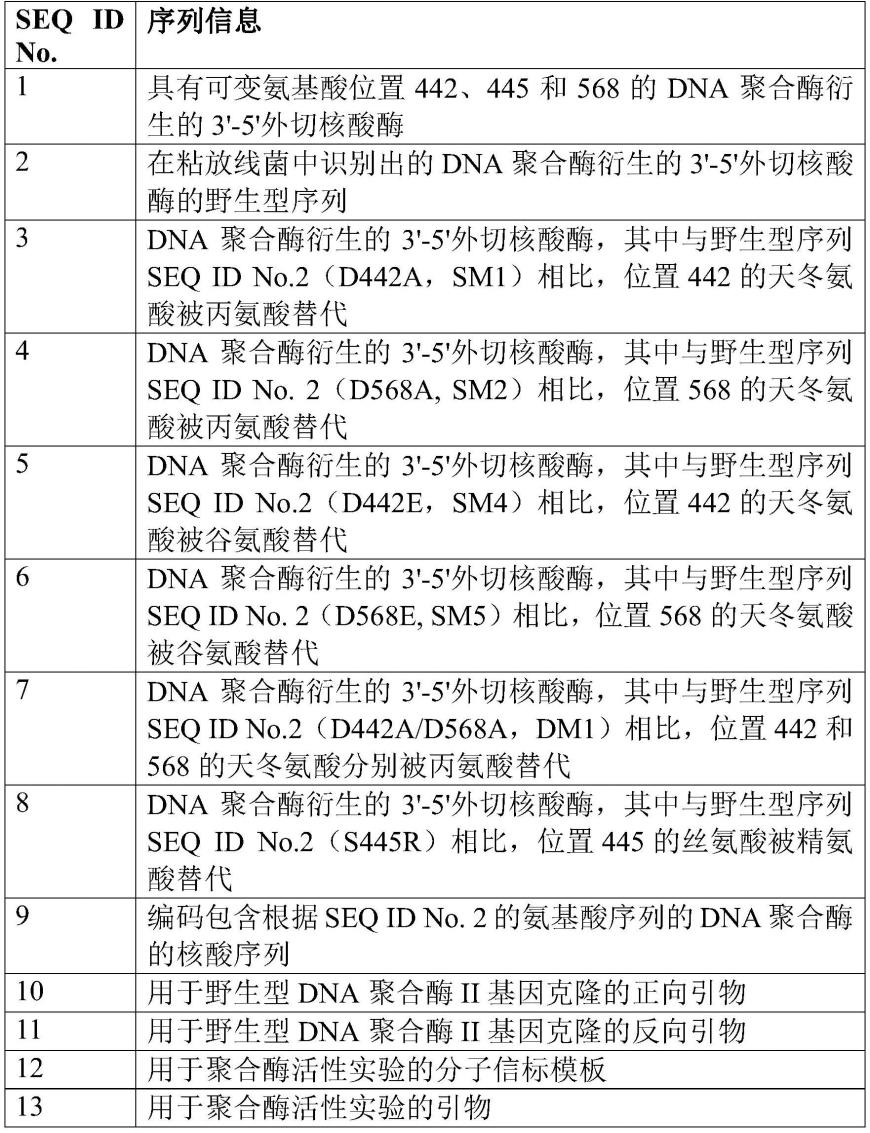
说明书和序列表中提到的序列号概述
[0208]
[0209]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。