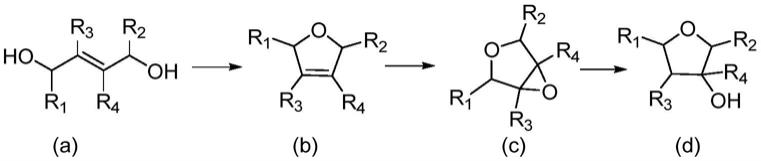
1.本发明属于化工技术领域,涉及一种3-羟基呋喃的制备方法,特别涉及1,4-丁烯二醇通过系列多活性组分催化剂催化选择性脱水、环氧化、加氢制备3-羟基四氢呋喃的方法。
背景技术:
2.3-羟基四氢呋喃是一种重要的医药化工中间体,在抗艾滋病药、抗癌药、降糖药等药物的生产中应用广泛。3-羟基四氢呋喃可以通过一些公知的技术,进一步转化为可以合成药物直接使用的手性3-羟基四氢呋喃。(cn201510572955.9,j.am.chem.soc.2012,acs catal.,2016,1598;febs journal 2013,280,3084-3093)。目前,3-羟基四氢呋喃主要用化学法合成,如用苹果酸(苹果酸酯、苹果酸还原产物1,2,4-丁三醇)和酒石酸(酒石酸酯)为起始原料通过酯化、还原、脱水环合合成3-羟基四氢呋喃及其手性体(s)-3-羟基四氢呋喃及(r)-3-羟基四氢呋喃(j.am.chem.soc.,1958,80,364;cn101367780a,cn104478833a,j.org.chem.,1983,48,2767;us2011/118511)。
3.李勇智等报道了以(s)-4-氯-3-羟基丁酸乙酯为原料,经过还原和环化两步反应得到目标产物(s)-3-羟基四氢呋喃,反应总收率为75.2%(应用化工,2008,037,191)。asim bhaumik等报道了一种钛硅分子筛催化丁烯醇环氧化耦合环合制备3-羟基四氢呋喃的方法,取得了较好的效果(chem commun,1998 463)。然而,丁烯醇难以廉价获得,且丁烯醇的性质及其活泼,副产物也较多。bats等报道了一种利用2-环氧乙烷基乙醇环合制备3-羟基四氢呋喃的方法,产物中同时存在大量的2-羟甲基氧杂环丁烷(tetrahedron,1982,38,2139),该方法所用的原料难以制备。王剑峰等报道了用小分子催化不对称合成(s)-3-羟基四氢呋喃,以4-氯丁醛和亚硝基苯为原料,经氨氧化反应、硼氢化钠还原、分子内环合制备(王剑峰,安普那韦中间体的合成工艺研究[d].浙江工业大学,2011.)。
[0004]
以二氢呋喃为原料,通过硅氢化或者硼氢化还原方法也可制得3-羟基四氢呋喃。brown等用手性硼催化剂实现了2,3-二氢呋喃和2,5-二氢呋喃的不对称硼氢化还原,产物3-羟基四氢呋喃的收率达到92%(j am chem soc,1986,108,2049)。hayashi等报道了以2,5-二氢呋喃为原料通过不对称硅氢化合成3-羟基四氢呋喃的方法(tetrahedron lett,1993,34,2335)。但由于催化剂难制备以及反应条件苛刻等原因,该路线在应用上受到一定限制。
技术实现要素:
[0005]
针对上述现有技术存在的问题,本发明的一个目的在于提供一种呋喃化合物(d)的制备方法,其特征在于,通过化合物(a)(取代或未取代的1,4-丁烯二醇,例如1,4-丁烯二醇)合成3-羟基呋喃的方法,所述制备方法如下反应式所示,包括以下步骤:
[0006][0007]
1)以化合物(a)为起始反应原料,在催化剂i存在下,于150-400℃,特别是250-300℃,通过间歇或者连续方式脱水环化反应例如0.1-24小时,得到化合物(b)(例如2,5-二氢呋喃);其中,r1、r2、r3、r4可各自独立地选自氢,羟基,氨基,巯基,c1-c10的烷基;
[0008]
2)步骤1)得到的化合物(b)在环氧化催化剂ii和溶剂的存在下与选自过氧化氢、过氧化氢水溶液、叔丁基过氧化氢、二甲基双环氧乙烷中的氧化物料,于-30-120℃下反应例如0.5-48h,得到化合物(c)(例如3,4-环氧四氢呋喃);
[0009]
3)步骤2)得到的化合物(c)在氢解催化剂iii和溶剂的存在下,于60-250℃,0.5-8mpa氢气压力下下反应例如0.5-24h,得到化合物(d)(取代或未取代的3-羟基四氢呋喃,例如3-羟基四氢呋喃)。
[0010]
根据本发明所述的方法,其中,所述步骤1)使用的催化剂i为载体负载的磷钨酸材料,所述的载体选自二氧化硅、三氧化二铝、二氧化锆、氧化铌、二氧化钛、硅铝分子筛中的一种或多种,其中磷钨酸相对于载体材料的重量百分比为0.1-1.0%,例如可以为0.1%,0.3%,0.5%,0.6%,0.8%等。
[0011]
根据本发明所述的磷钨酸的结构式为h3pw
12o40
,例如催化剂i可为0.6%h3o
40
pw
12
/al2o3。
[0012]
根据本发明所述的方法,其中,所述步骤2)中使用的催化剂ii为ti-si分子筛或者负载型或非负载型磷钨酸,优选二氧化硅或三氧化二铝负载的磷钨酸,例如磷钨酸相对于载体材料的重量百分比为0.1-1.0%,例如可以为0.1%,0.3%,0.5%,0.6%,0.8%等,如催化剂ii可以为0.6%h3o
40
pw
12
/al2o3。
[0013]
根据本发明所述的方法,其中,所述步骤2)中使用的溶剂可为选自水、二氯甲烷、二氯乙烷、醋酸、甲醇、乙醇、异丙醇、四氢呋喃、二氧六环、环己烷、甲苯中的一种或多种的混合物,优选为选自水、四氢呋喃、二氧六环中的一种或多种。
[0014]
根据本发明所述的方法,其中,所述步骤2)中使用的氧化物料优选过氧化氢或过氧化氢水溶液,例如30%双氧水。
[0015]
根据本发明所述的方法,其中,所述步骤3)中使用的催化剂iii为(0.1%-30%)a-(1%-20%)b/c,其中a为第一金属,其选自铼(re)、钼(mo)、锰(mn)、钴(co)、钨(w)中的一种或多种,b为第二金属,其选自镍(ni)、钌(ru)、铜(cu)、铂(pt)、钯(pd)、铑(rh)中的一种或多种;c为载体,其选自zno、mgo、al2o3、sio2、ceo2或者活性炭中的一种或多种。
[0016]
b为催化剂的主要活性组分,对反应起到关键作用,a为催化剂的助剂组分,主要是协同主催化组分催化加氢;催化剂载体起到担载活性组分和助剂组分的作用,使更多催化活性位点分散和暴露,提高催化剂的原子利用率。
[0017]
根据本发明,当各成分可选多种时,所选多种成分可以任意比例混合。
[0018]
催化剂iii可通过包括如下步骤的方法制备:通过浸渍法、沉积沉淀法、共沉淀法或者溶胶凝胶法先引入第一金属,其中第一金属占催化剂总量的质量分数为0.1%-30%,
然后引入第二金属,其中第二金属占催化剂总量的质量分数为1%-20%。
[0019]
优选地,根据本发明,催化剂iii优选为选自mo-pd、co-pd、w-rh的第一金属-第二金属负载于活性炭或者氧化铝上的二元负载型催化剂,例如20%mo-5%pd/c,20%w-5%rh/c。
[0020]
根据本发明所述的方法,其中,所述步骤3)中的反应温度可为80-200℃或90-150℃。
[0021]
根据本发明所述的方法,其中,所述步骤3)中使用的溶剂可为选自水、甲醇、乙醇、异丙醇、四氢呋喃、二氧六环、乙酸乙酯中的一种或者多种的混合物。
[0022]
根据本发明所述的方法,其中,步骤2)反应后滤除催化剂ii,直接补入催化剂iii进行步骤3)。
[0023]
根据本发明所述的方法,其中,化合物(a)为1,4-丁烯二醇,(d)为3-羟基四氢呋喃,所述制备方法如下反应式所示,
[0024][0025]
有益效果
[0026]
根据本发明的方法主要采用非均相催化反应,操作简单,催化剂价格低廉,分离也较为简单,有利于大规模工业化生产。特别是采用1,4-丁烯二醇为起始原料制备3-羟基四氢呋喃,相比于现行的1,2,4-丁三醇原料更加经济。
具体实施方式
[0027]
以下实施例仅是作为本发明的实施方案的例子列举,并不对本发明构成任何限制,本领域技术人员可以理解在不偏离本发明的实质和构思的范围内的修改均落入本发明的保护范围。
[0028]
制备实施例
[0029]
催化剂制备实施例1
[0030]
0.6%h3o
40
pw
12
/al2o3的制备:称取100g al2o3球(3-5mm,购自山东铝业公司)于110℃干燥过夜,450℃焙烧6h后,放入成滚圆机中慢速滚动,从喷头喷洒浓度为0.5mol/l的h3o
40
pw
12
.xh2o水溶液,同时辅助120℃热空气干燥,直到杂多酸h3o
40
pw
12
.xh2o用量大约占催化剂重量0.6wt%停止喷液,继续鼓风干燥2h后至于马弗炉中在400℃焙烧8h,催化剂标记为0.6%h3o
40
pw
12
/al2o3(负载量以载体计)。
[0031]
催化剂制备实施例2
[0032]
20%mo-5%pd/c催化剂的制备:(1)称取200g经过硝酸洗涤干燥的活性炭粉末(200-300目)置于氮气气氛中在500℃下焙烧12h,将浓度为0.2mol/l(以mo计)的七钼酸铵水溶液2l慢慢导入活性炭粉体中,充分搅拌。减压蒸干水分后将浆料在110℃下干燥,在450℃干燥8小时。(2)将17g氯化钯中加入25ml浓盐酸,慢慢加热使氯化钯溶解,然后加水稀释至200ml,并调ph至3.5,然后将氯化钯液体喷雾到上述炭粉末中,50℃缓慢干燥,然后在110℃氮气气氛中干燥10h后用5%氢气/氮气在350℃还原5h,经氮气气氛中降温后保存即可制得20%mo-5%pd/c催化剂。
[0033]
催化剂制备实施例3
[0034]
20%w-5%rh/c的制备:(1)称取200g经过硝酸洗涤干燥的活性炭粉末(200-300目)置于氮气气氛中在500℃下焙烧12h,将浓度为0.11mol/l(以w计)的偏钨酸铵水溶液2l慢慢导入活性炭粉体中,充分搅拌。减压蒸干水分后将浆料在110℃下干燥,在500℃干燥8小时。(2)将25.9g氯化铑加水稀释至200ml,然后将氯化铑液体喷雾到上述炭粉末中,50℃缓慢干燥,然后在110℃氮气气氛中干燥10h后用5%氢气/氮气在280℃还原5h,经氮气气氛中降温后保存即可制得20%w-5%rh/c催化剂。
[0035]
实施例
[0036]
实施例1
[0037]
在8mm
×
400mm固定床反应管上装填充12g载量为0.6%h3o
40
pw
12
/al2o3,将1,4-丁烯二醇预热至280℃通过催化剂床层,控制1,4-丁烯二醇物料的空速为0.5h-1
,同时n2吹扫流量为50ml/min,出料冷凝液体含有95.1%2,5-二氢呋喃,其他剩余组分主要为h2o和少量其他副产物;将收集到的500ml原料液加入5l反应釜中,加入500ml甲醇和50g ti-si分子筛(上海阿拉丁生化科技股份有限公司,比表面积350-450
㎡
/g),在80℃下慢慢滴入30%双氧水750ml,控制反应釜温度不超过85℃,反应后液体过滤催化剂后,直接补入20g 20%mo-5%pd/c催化剂,通入氮气气流,并搅拌0.5小时充分分解少量未反应的双氧水,然后用氮气置换三次釜内空气,然后充入5mpa氢气,在90℃反应12小时。
[0038]
在本发明中,产物用气相色谱分析,通过产物在色谱上的保留时间对产物进行定性,该分析是在配备了自动进样器aoc-20的岛津2010plus气相色谱上进行。气相色谱定量条件:色谱柱选用cp-wax 58(ffap,25m
×
0.25mm
×
0.2μm,chrompack);汽化室温度250℃(分流比1:30);fid检测器温度280℃;柱温箱温度在60℃保持1min,然后以20℃/min的速率升至250℃并保持5min;气路控制:n2 1ml/min(column),h2 30ml/min,空气300ml/min,尾吹n2 29ml/min。
[0039][0040]
气相色谱分析显示3-羟基四氢呋喃收率达到58.5%,副产物主要为脱水赤藓糖醇和3-甲氧基-4-羟基四氢呋喃。
[0041]
实施例2
[0042]
在8mm
×
400mm固定床反应管上装填充12g载量为0.6%h3o
40
pw
12
/al2o3,将1,4-丁烯二醇预热至280℃通过催化剂床层,控制液体物料的空速为0.8h-1
,同时n2吹扫流量为50ml/min,出料冷凝液体含有96.2%2,5-二氢呋喃,其他剩余组分主要为h2o和少量他副产物;将收集到的500ml原料液加入5l反应釜中,加入500ml甲醇和10g磷钨酸水合物(上海阿拉丁生化科技股份有限公司版,h3o
40
pw
12
.xh2o),在80℃下慢慢滴入30%双氧水750ml,控制反应釜温度不超过85℃,反应后液体过滤催化剂后,直接补入20g 20%mo-5%pd/c催化剂,通入氮气气流,并搅拌0.5小时充分分解少量未反应的双氧水,然后用氮气置换三次釜内空气,然后充入5mpa氢气,并不断补充气体保持体系压力在4.5-5.0mpa,在90℃反应12小时,色谱分析显示3-羟基四氢呋喃收率达到75.2%。
[0043]
实施例3
[0044]
具体实施过程同实施例1,所不同的是环氧化催化剂为负载型固体酸催化剂0.6%
h3o
40
pw
12
/al2o3,3-羟基四氢呋喃收率达82.0%。
[0045]
实施例4
[0046]
具体实施过程同实施例3,所不同的是环氧反应后产品经过二氯乙烷萃取纯化后投入后续加氢反应,加氢步骤中所使用的溶剂为无水四氢呋喃,3-羟基四氢呋喃相对于环氧四氢呋喃收率达86.0%,相对于原料收率可达68%,产品中无醇解开环产产物和脱水赤藓糖醇。
[0047]
实施例5
[0048]
具体实施过程同实施例4,所不同的是加氢催化剂选择20%w-5%rh/c,3-羟基四氢呋喃相对于环氧四氢呋喃收率达92.0%,产品中无醇解开环产物和脱水赤藓糖醇。
[0049]
实施例6
[0050]
具体实施过程同实施例4,所不同的是加氢催化剂选择5%pd/c(购自陕西瑞科新材料股份有限公司),3-羟基四氢呋喃相对于环氧四氢呋喃收率达46.0%,原料未反应完全。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。