1.本发明涉及药物化学技术领域,特别涉及蟾酥甾二烯衍生物及其制备方法和应用。
背景技术:
2.癌症严重威胁人类健康,2018年世界卫生组织旗下的international agency for research on cancer(iarc)发布的全球癌症报告中指出:在刚刚过去的这一年,有1810万人确诊为癌症患者,有960万人因癌症丧生,而在我国有将近360万人确诊为癌症患者。
3.蟾酥是我国一味名贵传统中药,其中的蟾酥甾二烯是其主要的活性成分,具有显著地抗肿瘤活性,因此这类化合物具有用于制备抗肿瘤药物的潜能。然而水溶极低、毒性强是蟾酥甾二烯类化合物所存在的最主要的问题,急需对其水溶性和毒性问题进行改进。
技术实现要素:
4.基于此,有必要提供一种涉及蟾酥甾二烯衍生物及其制备方法和应用。该类蟾酥甾二烯衍生物的水溶性得到提高、毒性降低,同时保持了抗肿瘤的活性,可用作防治恶性肿瘤的药物。
5.一种蟾酥甾二烯衍生物或其药学上可接受的盐,述蟾酥甾二烯衍生物具有式(i)所示结构:
[0006][0007]
其中,a为-(crarb)
n-;
[0008]
n为0、1、2、3、4、5、6、7、8、9或10;
[0009]
r1为羟基、r2为氢,或r1与r2共同形成环氧基;
[0010]
r3为氢或c
1-6
烷基酰氧基;
[0011]
r4、r5、r6和r7各自独立地为:氢、c
1-6
烷基、3-8元环烷基或5-8元杂环烷基;或r4与r6共同形成取代或未取代的5-8元含氮杂环烷基;或r4与r7共同形成取代或未取代的5-8元含氮杂环烷基;或r5与r6共同形成取代或未取代的5-8元环烷基;或r5与r7共同形成取代或未取代的5-8元含氮杂环烷基;
[0012]
ra和rb各自独立地为:氢、c
1-6
烷基或3-8元环烷基。
[0013]
上述蟾酥甾二烯衍生物的制备方法,包括以下步骤:
[0014][0015]
将式(i-1)所述化合物和进行反应,制得式(i-3)所述化合物;
[0016]
将式(i-3)所述化合物和式(i-4)所述化合物进行缩合反应,制得式(i)所述化合物。
[0017]
一种药物组合物,包括
[0018]
1)上述蟾酥甾二烯衍生物和其药学上可接受的盐中至少一种,
[0019]
2)药学上可接受载体、赋形剂、佐剂、辅料和稀释剂中至少一种。
[0020]
上述蟾酥甾二烯衍生物或其药学上可接受的盐、或上述药物组合物在制备治疗或预防肿瘤的药物中的应用。
[0021]
本发明创新性地利用蟾酥甾二烯的抗肿瘤活性和亲水性的苦参酸相结合策略,通过linker将这两者链接起来,获得系列结构新颖的蟾酥甾二烯衍生物,不仅水溶性得到了提高,活性得到了保持,毒性却下降了。故上述蟾酥甾二烯衍生物可以解决蟾酥甾二烯衍生物由于毒性合类药性问题,具有良好的应用前景。
具体实施方式
[0022]
为了便于理解本发明,下面将对本发明进行更全面的描述,并给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
[0023]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相
关的所列项目的任意的和所有的组合。
[0024]
术语解释
[0025]
除非特别注明,本发明中所用的术语具有如下定义:
[0026]
本发明中的若化合物存在立体异构体,在没有特别指明时,应理解为包括r构型、s构型以及消旋体。
[0027]
本发明中所述的“取代”表示被一个或多个基团所替代。当多个基团从同一系列候选取代基中选择时,它们可以相同,也可以不同。
[0028]
本发明中所述的“任选地”表示所定义基团可从一系列候选基团中进行选择,也可以不选。
[0029]
本发明中所述的“取代或未取代”表示所定义的基团可以被取代,也可以不被取代。当所定义的基团被取代时,应理解为任选被本领域可接受的基团所取代,包括但不限于:c
1-6
烷基、含有3-8个环原子的环烷基、含有4-9个环原子的杂环烷基、硅烷基、羰基、羟基或-nrr
′
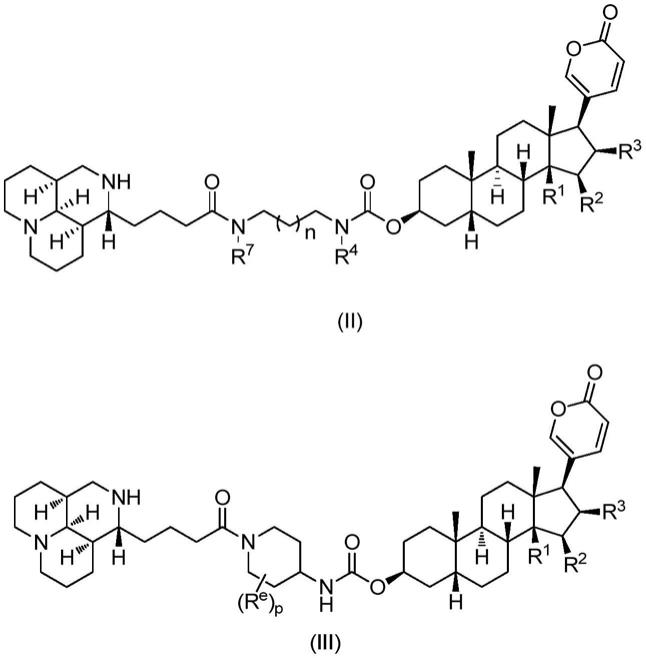
,且上述基团也可以进一步被本领域可接受取代基取代;可理解的,-nrr
′
中的r和r
′
各自独立地为本领域可接受的基团所取代,包括但不限于h、c
1-6
烷基,且r和r
′
可和与r、r
′
相连的n一起形成5-10元杂环烷基;所述c
1-6
烷基、含有3-8个环原子的环烷基、含有4-9个环原子的杂环烷基任选进一步被一个或多个以下基团取代:c
1-6
烷基、含有3-8个环原子的环烷基、含有3-8个环原子的杂环基、卤素、羟基、硝基或氨基。
[0030]
本发明中所述的“烷基”表示特定原子个数下的饱和的直链和支链烷基,所述c
1-6
烷基是指含有1至6个碳原子的烷基。非限定性实施例包括:甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、1-乙基丙基、2-甲基丁基、3-甲基丁基、正己基、1-乙基-2-甲基丙基、1,1,2-三甲基丙基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2-乙基丁基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2,3-二甲基丁基。
[0031]“环烷基”指饱和或部分不饱和单环或多环环状烃基取代基。3-8元环烷基是指包括3至8个碳原子。非限定性实施例包括:环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环己二烯基、环庚基、环庚三烯基、环辛基等。
[0032]“含氮杂环烷基”指至少含有一个氮原子的饱和或部分不饱和单环或多环环状烃基取代基。5-8元含氮杂环烷基是指包括5至8个原子(碳原子和氮原子总和)。非限定性实施例包括:四氢吡咯基、四氢咪唑基、四氢吡唑基、哌啶基、哌嗪基、六氢嘧啶、六氢哒嗪、环己亚胺基、1,2-高哌嗪基、1,3-高哌嗪基、1,4-高哌嗪基、1,2-二氮杂八环基、1,3-二氮杂八环基、1,4-二氮杂八环基、1,5-二氮杂八环基、四氢吡啶基等。
[0033]“药学上可接受的盐”是指所示结构中的任一化合物与酸或碱所形成的适合用作药物的盐。药学上可接受的盐包括无机盐和有机盐。其中,一类盐是本发明化合物与酸形成的盐。适合形成盐的酸包括但并不限于:盐酸、氢溴酸、氢氟酸、硫酸、硝酸、磷酸等无机酸;甲酸、乙酸、三氟乙酸、丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、酒石酸、柠檬酸、苦味酸、苯甲酸、甲磺酸、乙磺酸、对甲苯磺酸、苯磺酸、萘磺酸等有机酸;以及脯氨酸、苯丙氨酸、天冬氨酸、谷氨酸等氨基酸。另一类盐是本发明化合物与碱形成的盐,适合形成盐的碱包括但并不限于:碱金属盐(例如钠盐或钾盐)、碱土金属盐(例如镁盐或钙盐)、铵盐(如低级的烷醇铵盐以及其它药学上可接受的胺盐),例如甲胺盐、乙胺盐、丙胺盐、二甲
基胺盐、三甲基胺盐、二乙基胺盐、三乙基胺盐、叔丁基胺盐、乙二胺盐、羟乙胺盐、二羟乙胺盐、三羟乙胺盐,以及分别由吗啉、哌嗪、赖氨酸形成的胺盐。
[0034]
使用方法
[0035]
利用本发明所得的蟾酥甾二烯衍生物可给药于人,可以口服、直肠、肠胃外(静脉内、肌肉内或皮下)、膀胱灌注、局部给药(粉剂、软膏剂或滴剂)。
[0036]
用于口服给药的固体剂型包括胶囊剂、片剂、丸剂、散剂和颗粒剂。在这些固体剂型中,活性化合物与至少一种常规惰性赋形剂(或载体)混合,如柠檬酸钠或磷酸二钙,或与下述成分混合:(a)填料或增容剂,例如,淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸;(b)粘合剂,例如,羟甲基纤维素、藻酸盐、明胶、聚乙烯基吡咯烷酮、蔗糖和阿拉伯胶;(c)保湿剂,例如,甘油;(d)崩解剂,例如,琼脂、碳酸钙、马铃薯淀粉或木薯淀粉、藻酸、某些复合硅酸盐、和碳酸钠;(e)缓溶剂,例如石蜡;(f)吸收加速剂,例如,季胺化合物;(g)润湿剂,例如鲸蜡醇和单硬脂酸甘油酯;(h)吸附剂,例如,高岭土;和(i)润滑剂,例如,滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠,或其混合物。胶囊剂、片剂和丸剂中,剂型也可包含缓冲剂。
[0037]
固体剂型如片剂、糖丸、胶囊剂、丸剂和颗粒剂可采用包衣和壳材制备,如肠衣和其它本领域公知的材料。它们可包含不透明剂,并且,这种组合物中活性化合物或化合物的释放可以延迟的方式在消化道内的某一部分中释放。可采用的包埋组分的实例是聚合物质和蜡类物质。必要时,活性化合物也可与上述赋形剂中的一种或多种形成微胶囊形式。
[0038]
用于口服给药的液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆或酊剂。除了活性化合物外,液体剂型可包含本领域中常规采用的惰性稀释剂,如水或其它溶剂,增溶剂和乳化剂,例知,乙醇、异丙醇、碳酸乙酯、乙酸乙酯、丙二醇、1,3-丁二醇、二甲基甲酰胺以及油,特别是棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油或这些物质的混合物等。
[0039]
除了这些惰性稀释剂外,上述组合物也可包含助剂,如润湿剂、乳化剂和悬浮剂、甜味剂、矫味剂和香料。
[0040]
除了活性化合物外,悬浮液可包含悬浮剂,例如,乙氧基化异十八烷醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、甲醇铝和琼脂或这些物质的混合物等。
[0041]
用于肠胃外注射或膀胱灌注的组合物可包含生理上可接受的无菌含水或无水溶液、分散液、悬浮液或乳液,和用于重新溶解成无菌的可注射溶液或分散液的无菌粉末。适宜的含水和非水载体、稀释剂、溶剂或赋形剂包括水、乙醇、多元醇及其适宜的混合物。
[0042]
用于局部给药的本发明化合物的剂型包括软膏剂、散剂、喷射剂和吸入剂。活性成分在无菌条件下与生理上可接受的载体及任何防腐剂、缓冲剂,或必要时可能需要的推进剂一起混合。
[0043]
详细解释
[0044]
本发明一实施方式提供了一种蟾酥甾二烯衍生物或其药学上可接受的盐,具有式(i)所示结构:
[0045][0046]
其中,其中,a为-(crarb)
n-;
[0047]
n为0、1、2、3、4、5、6、7、8、9或10;
[0048]
r1为羟基、r2为氢,或r1与r2共同形成环氧基;
[0049]
r3为氢或c
1-6
烷基酰氧基;
[0050]
r4、r5、r6和r7各自独立地为:氢、c
1-6
烷基、3-8元环烷基或5-8元杂环烷基;或r4与r6共同形成取代或未取代的5-8元含氮杂环烷基;或r4与r7共同形成取代或未取代的5-8元含氮杂环烷基;或r5与r6共同形成取代或未取代的5-8元环烷基;或r5与r7共同形成取代或未取代的5-8元含氮杂环烷基;
[0051]
ra和rb各自独立地为:氢、c
1-6
烷基或3-8元环烷基。
[0052]
在一实施例中,r3为氢或c
1-4
烷基酰氧基;进一步地,r3为氢或乙酰氧基;
[0053]
在一实施例中,r1与r2共同形成环氧基、r3为乙酰氧基。
[0054]
在一实施例中,r1为羟基、r2和r3均为氢。
[0055]
在一实施例中,ra和rb各自独立地为:氢、c
1-4
烷基或3-6元环烷基。
[0056]
在一实施例中,a为-(crarb)
n-;进一步地,ra和rb为h;进一步地,n为0、1、2、3、4或5。可理解的,当n为0,表示a不存在,式(i)具有以下结构:
[0057][0058]
进一步地,r4、r5、r6和r7各自独立地为:氢、c
1-4
烷基、3-6元环烷基或5-6元杂环烷基;或r4与r6共同形成取代或未取代的5、6或7元含氮杂环烷基;或r4与r7共同形成取代或未取代的5、6或7元含氮杂环烷基;或r5与r6共同形成取代或未取代的5、6或7元环烷基;或r5与r7共同形成取代或未取代的5、6或7元含氮杂环烷基。
[0059]
在一实施例中,r4和r7为h;在一实施例中,r5和r6为h;在一实施例中,r4与r6共同形成取代或未取代的6元含氮杂环烷基;在一实施例中,r4与r7共同形成取代或未取代的6元含氮杂环烷基;在一实施例中,r5与r7共同形成取代或未取代的6元含氮杂环烷基;
[0060]
进一步地,5-8元环烷基或所述5-8元含氮杂环烷基进一步被至少一个r取代,r选自:-h、c
1-6
烷基、3-8元环烷基、羟基、羧基、羰基或氧代基。
[0061]
进一步地,含氮杂环烷基选自:哌啶基、哌嗪基、六氢嘧啶、六氢哒嗪、1,2-高哌嗪基、1,3-高哌嗪基、或1,4-高哌嗪基;
[0062]
进一步地,环烷基选自:环戊基或环己基;
[0063]
进一步地,上述蟾酥甾二烯衍生物具有式(ii)或式(iii)所示结构:
[0064][0065]
re选自h、c
1-6
烷基或3-8元环烷基;
[0066]
p为1、2、3、4、5、6、7或8。
[0067]
进一步地,上述蟾酥甾二烯衍生物选自如下任一化合物:
[0068]
[0069][0070]
进一步地,上述蟾酥甾二烯衍生物的药学上可接受的盐为有机盐;更进一步地,上述蟾酥甾二烯衍生物的药学上可接受的盐为蟾酥甾二烯衍生物的草酸盐、丙二酸盐、琥珀酸盐、富马酸盐、马来酸盐、乳酸盐、苹果酸盐、酒石酸盐、柠檬酸盐、或苦味酸盐;更进一步地,为上述蟾酥甾二烯衍生物的药学上可接受的盐为蟾酥甾二烯衍生物的酒石酸盐。
[0071]
本发明还提供了上述蟾酥甾二烯衍生物的制备方法,包括以下步骤:
[0072]
s101:将式(i-1)所述化合物和进行反应,制得式(i-3)所述化合物;
[0073][0074]
进一步地,优选步骤s101包括以下步骤:
[0075]
s1011:将式(i-1)所述化合物和4-硝基氯甲酸苯酯进行反应,制得式(i-2)所述化合物;
[0076][0077]
进一步地,步骤s1011包括以下步骤:使式(i-1)所示结构的化合物、4-硝基氯甲酸苯酯、碱和溶剂混合,进行反应,后处理,制得式(i-2)所述化合物;
[0078]
更进一步地,所述碱为三乙胺、吡啶、二异丙基乙基胺和4-(n,n-二甲基)氨基吡啶中的一种或多种;更进一步地,碱为吡啶;
[0079]
更进一步地,所述溶剂选自:二氯甲烷、氯仿、四氢呋喃和乙腈中的一种或多种;更进一步地,溶剂为二氯甲烷;
[0080]
更进一步地,反应温度为15~45℃;
[0081]
更进一步地,后处理方法为:反应结束后,采用饱和碳酸钠或饱和碳酸钾溶液反复洗涤,然后再用饱和食盐水洗涤,干燥、浓缩。
[0082]
s1012:将式(i-2)所述化合物和进行反应,制得式(i-3)所述化合物;
[0083]
[0084]
进一步地,步骤s1012包括以下步骤:将式(i-2)所述化合物、碱和溶剂混合,进行反应,反应完成后进行后处理,得到式(i-3)所述化合物;
[0085]
更进一步地,所述碱为三乙胺、吡啶、二异丙基乙基胺和4-(n,n-二甲基)氨基吡啶中的一种或多种;更进一步地,碱为三乙胺;
[0086]
更进一步地,所述溶剂选自:二氯甲烷、氯仿、四氢呋喃和乙腈中的一种或多种;更进一步地,溶剂为二氯甲烷。
[0087]
s102:将式(i-3)所述化合物和式(i-4)所述化合物进行缩合反应,制得式(i)所述化合物。
[0088][0089]
进一步地,步骤s102中将式(i-3)所述化合物中的n原子上保护基后再进行后续反应,可以采用本领域常见的nh保护基,包括但不限于:-boc、-cbz、-ac、-ts、-ms、-bz、-bn、-pmb、或schiff碱;优选采用-boc,具有更高的产率,且反应条件较为温和;
[0090]
进一步地,步骤s102包括以下步骤:
[0091]
s1021:将式(i-4)所述化合物上boc保护基,制备式(i-4')所述化合物;
[0092][0093]
可理解的,可以直接市购含有boc保护基的原料,此时可以省略步骤s1021,不应理解为对本发明的限制。
[0094]
s1022:式(i-3)所述化合物与式(i-4')所述化合物进行反应,制得式(i')所述化合物;
[0095][0096]
进一步地,步骤s1022包括以下步骤:将式(i-3)所示结构化合物和式(i-4')所示结构化合物、缩合试剂、碱和溶剂混合,进行反应,制得式(i')所述化合物;
[0097]
进一步地,缩合试剂为edci、dcc和hobt/edci中的一种或多种;更进一步地,缩合试剂为edci;
[0098]
进一步地,碱为三乙胺、吡啶、二异丙基乙基胺和4-(n,n-二甲基)氨基吡啶中的一种或多种;更进一步地,碱为三乙胺
[0099]
进一步地,溶剂选自:二氯甲烷、氯仿、四氢呋喃和乙腈中的一种或多种;更进一步地,溶剂为二氯甲烷;
[0100]
进一步地,温度为15~45℃;
[0101]
s1023:脱除式(i')所述化合物的boc保护基,制得式(i)所述化合物;
[0102][0103]
进一步地,步骤s1023包括以下步骤:将式(i')所示结构化合物、酸和溶剂混合,进行反应;
[0104]
进一步地,酸为盐酸、硫酸、对甲苯磺酸和三氟乙酸中的一种或多种;更进一步地,酸为对甲苯磺酸和三氟乙酸;更进一步地,酸为对甲苯磺酸。
[0105]
进一步地,反应温度为15~45℃;
[0106]
本发明还提供了一种药物组合物,包括
[0107]
1)上述蟾酥甾二烯衍生物和其药学上可接受的盐中至少一种;
[0108]
2)药学上可接受载体、赋形剂、佐剂、辅料和稀释剂中的至少一种。
[0109]
本发明还提供了上述蟾酥甾二烯衍生物或其药学上可接受的盐、或上述药物组合物在制备治疗或预防肿瘤的药物中的应用。
[0110]
进一步地,肿瘤为肺癌、宫颈癌、肝癌、乳腺癌、血癌、胰腺癌、胃癌、食道癌、直肠癌、膀胱癌或肾癌。
[0111]
本发明还提供了一种治疗或预防肿瘤的方法,包括施加治疗有效量的上述蟾酥甾二烯衍生物或其药学上可接受的盐,或上述药物组合物。
[0112]
制备实施例
[0113]
下面结合具体实施例对本发明作进一步阐述,但本发明不局限于此。
[0114]
下述制备例中,1h-nmr用varian mercury amx 500型仪器测定。ms用vg zab-hs或vg-7070型以及esquire 3000plus-01005测定。所有溶剂在使用前均经过重新蒸馏,所使用的无水溶剂均是按标准方法干燥处理获得。除另有说明外,所有反应均是在氩气保护下进行并用tlc跟踪,后处理时均经饱和食盐水洗和无水硫酸镁干燥过程。产品的纯化除另有说明外均使用硅胶的柱色谱法,所使用的硅胶为200-300目,gf
254
为青岛海洋化工厂或烟台缘博硅胶公司生产。
[0115]
实施例1:化合物a1的合成
[0116][0117]
在50ml圆底烧瓶中,将氯甲酸对硝基苯酯(1.206g,6mmol)溶于20ml无水二氯甲烷中,加入干燥吡啶(0.67ml),搅拌下加入蟾毒灵(2mmol),室温下搅拌6小时,反应结束后用饱和碳酸钠洗5-8次,再用饱和食盐水洗2两次,用硫酸镁干燥。过滤除去硫酸镁后,再加入10ml无水二氯甲烷和三乙胺(0.5ml),再加入乙二胺(4mmol)室温搅拌1h。反应结束后用饱和碳酸钠洗5-8次,再用饱和食盐水洗2两次,用硫酸镁干燥。过滤后,依次加入n-boc苦参酸(1.46g,4mmol),三乙胺(6mmol),edci(6mmol,1.15g),室温反应3h,反应结束后用饱和碳酸钠洗3次,无水硫酸钠干燥,减压浓缩后溶于甲醇(20ml),加入对甲基苯磺酸(6mmol,1.03mg),室温搅拌120h,tlc检测原料反应完全后,加乙酸乙酯(40ml),依次用饱和碳酸钠洗1次,水洗2次,再用饱和食盐水洗2次,并用硫酸镁干燥。过滤除去硫酸镁并减压浓缩后,经柱层析(dcm:meoh:et3n=30:1:0.3,v/v/v)得白色粉末a1(534mg,37%)。1h nmr(500mhz,dmso-d6)δ7.95(d,j=9.4hz,1h),7.89(s,1h),7.53(s,1h),7.04(t,j=5.6hz,1h),6.30(d,j=9.7hz,1h),4.83(s,1h),4.27
–
4.20(m,1h),3.19
–
2.96(m,6h),2.71(dd,j=22.4,10.6hz,2h),2.55(dd,j=11.9,3.4hz,1h),2.47(t,j=7.0hz,1h),2.09
–
1.99(m,4h),1.91(t,j=13.8hz,1h),1.85
–
1.74(m,6h),1.69
–
1.54(m,7h),1.56
–
1.45(m,7h),1.44
–
1.27(m,9h),1.26
–
1.02(m,7h),0.91(s,3h),0.61(s,3h);esi-ms(m/z)721.6[m 1]
。
[0118]
实施例2:化合物a2的合成
[0119][0120]
应操作如a1的制备,原料用丙二胺代替乙二胺;经柱层析(dcm:meoh:et3n=30:1:0.3,v/v/v)梯度洗脱,得到目标化合物a2(588mg,40%)。1h nmr(500mhz,dmso-d6)δ7.93(dd,j=9.8,2.4hz,1h),7.78(t,j=5.3hz,1h),7.57
–
7.50(m,1h),6.95(t,j=5.5hz,1h),6.28(d,j=9.7hz,1h),4.80(s,1h),3.44(s,4h),3.11
–
2.87(m,6h),2.68(dd,j=20.0,11.0hz,2h),2.50
–
2.39(m,2h),2.07
–
1.96(m,4h),1.89(t,j=13.1hz,1h),1.83
–
1.72(m,5h),1.64
–
1.54(m,7h),1.53
–
1.44(m,8h),1.43
–
1.37(m,2h),1.35
–
1.21(m,7h),1.21
–
1.01(m,6h),0.88(s,3h),0.59(s,3h);esi-ms(m/z)735.6[m 1]
。
[0121]
实施例3:化合物a3的合成
[0122][0123]
应操作如a1的制备,原料用丁二胺代替乙二胺;经柱层析(dcm:meoh:et3n=30:1:0.3,v/v/v)梯度洗脱,得到目标化合物a3(524mg,35%)。1h nmr(500mhz,dmso-d6)δ7.95(dd,j=9.7,2.6hz,1h),7.82(t,j=5.6hz,1h),7.53(d,j=2.4hz,1h),6.98(t,j=5.5hz,1h),6.30(d,j=9.7hz,1h),4.86
–
4.77(m,1h),4.18(s,1h),3.18
–
3.06(m,3h),3.06
–
2.91(m,5h),2.76
–
2.67(m,2h),2.64(dd,j=12.1,4.1hz,1h),2.49
–
2.45(m,1h),2.09
–
1.98(m,5h),1.96
–
1.85(m,1h),1.84(dd,j=11.8,2.9hz,1h),1.82
–
1.75(m,4h),1.64
–
1.42(m,16h),1.39
–
1.30(m,10h),1.28
–
1.23(m,6h),0.89(s,3h),0.61(s,3h);esi-ms(m/z)749.6[m 1]
。
[0124]
实施例4:化合物a4的合成
[0125][0126]
应操作如a1的制备,原料用4-氨基哌啶代替乙二胺;经柱层析(dcm:meoh:et3n=30:1:0.3,v/v/v)梯度洗脱,得到目标化合物a4(700mg,46%)。1h nmr(500mhz,dmso-d6)δ7.94(dd,j=9.8,2.4hz,1h),7.75(d,j=7.7hz,1h),7.52(d,j=1.8hz,1h),6.29(d,j=9.7hz,1h),4.85(s,1h),4.16(s,1h),3.87(d,j=13.1hz,2h),3.78
–
3.70(m,1h),3.02
–
2.98(m,1h),2.92(t,j=8.3hz,2h),2.68(dd,j=21.3,11.0hz,2h),2.48
–
2.38(m,3h),2.13
–
1.99(m,4h),1.98
–
1.93(m,2h),1.84
–
1.73(m,6h),1.65
–
1.56(m,7h),1.56
–
1.43(m,8h),1.43
–
1.29(m,6h),1.28
–
1.15(m,9h),1.15
–
1.02(m,3h),0.89(s,3h),0.60(s,3h);esi-ms(m/z)761.6[m 1]
。
[0127]
实施例5:化合物a5的合成
[0128][0129]
应操作如a1的制备,原料用华蟾毒精代替蟾毒灵,用4-氨基哌啶代替乙二胺,脱boc时,用5%的三氟乙酸二氯甲烷溶液代替对甲苯磺酸;经柱层析(dcm:meoh:et3n=30:1:0.3,v/v/v)梯度洗脱,得到目标化合物a5(490mg,30%)。1h nmr(500mhz,dmso-d6)δ9.19(s,2h),8.02(s,1h),7.87(d,j=9.9hz,1h),7.49(d,j=2.5hz,1h),6.25(d,j=9.8hz,1h),5.49(d,j=9.2hz,1h),4.85(s,1h),3.96
–
3.82(m,2h),3.81
–
3.67(m,2h),3.23
–
3.14(m,1h),3.00
–
2.64(m,6h),2.18
–
2.04(m,3h),2.02
–
1.92(m,2h),1.83(s,3h),1.81
–
1.75(m,3h),1.76
–
1.61(m,7h),1.61
–
1.45(m,11h),1.43
–
1.29(m,6h),1.29
–
1.10(m,7h),1.06
–
0.99(m,1h),0.94(s,3h),0.72(s,3h);esi-ms(m/z)817.6[m 1]
。
[0130]
实施例6:化合物a4酒石酸盐的制备
[0131][0132]
将a4(1mmol)溶于30ml乙醇中,加入酒石酸(150mg,1mmol),室温搅拌溶解澄清后,减压浓缩后得到白色固体a4酒石酸盐。
[0133]
本发明所提及的化合物的有机酸和无机酸盐均可用类似方法进行制备。
[0134]
试验实施例1:体外抗肿瘤活性实验
[0135]
(1)试验材料
[0136]
人肺癌细胞a549、人宫颈癌细胞hela、人肝癌细胞hep3b、人乳腺癌细胞mcf-7、人急性淋巴细胞白血病t淋巴细胞ccrf-cem、人白血病细胞mv-4-11、腺癌细胞panc-1、人肝癌细胞hepg2、人肝癌细胞bel-7402。
[0137]
阳性对照为长春瑞滨(vinorelbine)(按常规方法配制);纯度由hplc-uv检测98%以上。待测化合物和阳性对照物以生理盐水稀释,浓度梯度为300nm、100nm、30nm、10nm、3.0nm、1.0nm、0.3nm、0.1nm。
[0138]
(2)试验方法
[0139]
srb还原法:
[0140]
根据细胞生长速率,将处于对数生长期的肿瘤细胞以100μl/孔接种于96孔培养板,贴壁生长24h再加待测化合物或阳性对照物10μl/孔。每个浓度设三复孔。并设相应浓度
的生理盐水溶媒对照及无细胞调零孔。肿瘤细胞在37℃、5%co2条件下培养72h,然后倾去培养液(rpmi-1640),用10%冷tca固定细胞,4℃放置1h后用蒸馏水洗涤5次,空气中自然干燥。然后加入由1%冰醋酸配制的srb(sigma)4mg/ml溶液100μl/孔,室温中染色15分钟,去上清液,用1%醋酸洗涤5次,空气干燥。最后加入150μl/孔的tris溶液,酶标仪515nm波长下测定a值。按以下列公式计算肿瘤细胞生长的抑制率:
[0141]
抑制率%=[(阴性对照吸光值
–
空白吸光值)
–
(样品吸光值
–
空白吸光值)]/(阴性对照吸光值
–
空白吸光值)
×
100%
[0142]
药物作用浓度:300nm、100nm、30nm、10nm、3.0nm、1.0nm、0.3nm、0.1nm。用graphpad prism 4拟合出ic
50
。
[0143]
表1、化合物a1-5对a549,hela和hep3b肿瘤细胞的抑制活性
[0144][0145]
通过对蟾酥甾二烯衍生物a1-a5对三种细胞株的细胞增殖抑制活性研究发现,本发明的化合物基本保持了抗肿瘤活性,其中a4化合物活性最佳(参见表1)。对a4又选取6株人源肿瘤细胞株,均显示了良好的抑制活性(参见表2)。
[0146]
表2、化合物a4对多种人源肿瘤细胞的抑制活性
[0147][0148]
相比于原型蟾毒灵,本发明的化合物活性得到保持,毒性显著降低和水溶性明显提高。蟾毒灵的ld
50
=2.2mg/kg,a4酒石酸盐的ld
50
=50mg/kg。蟾毒灵的水溶性小于1μg/ml,a4酒石酸盐的水溶性大于20mg/ml,提高了20000倍以上。
[0149]
对比实验
[0150][0151]
单端被乙酰化的产物b没有抗肿瘤活性,表明酰化会让化合物的活性丧失。同时,与boc保护的苦参酸酰化的产物c,尽管它也保留了含氮的水溶性基团,但是该化合物也是没有抗肿瘤活性。
[0152]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0153]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。