1.本发明属于化工技术领域,具体涉及一种哌嗪二酮衍生物及其制备方法。
背景技术:
2.哌嗪二酮类化合物(diketopiperazines,dkps)的特征结构是由两个氨基酸通过肽键缩合而成的环二肽(cyclic dipeptides),如下式所示。稳定的六元环骨架结构使dkps在药物化学中成为一个重要的药效闭,表现出了抗细菌、抗真菌、抗病毒、抗肿瘤、免疫抑制、神经保护、抗疟疾、抗朊病毒、抗高血糖等多种显著的生物活性和药理活性。
[0003][0004]
2,5-哌嗪二酮类化合物广泛存在于自然界中,不仅存在于真菌、真菌、植物以及哺乳动物的代谢产物中,也在一些食物中尤其是发酵食物中经由多肽降解而生成。随着研究的深入,研究人员发现2,5-哌嗪二酮类化合物不仅具有一系列利于各类受体结合的优势结构,同时也具有一些特别的特性使其在药物研发上具有显著优势。首先,结构简单,仅一个六元刚性环;其次,六个原子中四个位置可以进行拓展和立体构型控制。最后,对于蛋白质的水解作用是非常稳定的。近期的研究也显示了2,5-哌嗪二酮类化合物存在着广泛的、多样的生物活性。这一系列的特点都使得该类化合物逐步成为研究的热点之一。
[0005]
肿瘤免疫逃逸是肿瘤发生发展的重要环节,也是肿瘤细胞抵御淋巴细胞攻击的有效途径。肿瘤细胞可通过多种途径逃避免疫攻击,表达抑制性配体是其中一种途径。ctla-4是一种大家熟知的能够抑制淋巴细胞功能、诱导淋巴细胞凋亡,从而促进肿瘤发生的抑制性配体。除了ctla-4,pd-l1和pd-1的相互作用也可促进肿瘤免疫逃逸的发生。关于pd-1/pd-l1在肿瘤免疫逃逸中的机制研究已成为肿瘤免疫研究中的热点。肿瘤细胞表面表达的pd-l1与til表达的pd-1结合后,可引起pd-1胞质区itsm结构域中的酪氨酸残基发生磷酸化,通过募集shp-2磷酸酶,抑制下游信号通路的活化,抑制t细胞的功能,诱导til的凋亡,从而使得肿瘤细胞逃逸了免疫细胞的诱导凋亡作用。
[0006]
细胞程序性死亡受体1(programmed death-1,pd-1)是ishida等通过消减杂交技术于1992年发现的。pd-1主要表达于活化的t淋巴细胞、b淋巴细胞和巨噬细胞表面。程序性死亡配体-1(programmed death 1ligand 1,pd-l1),又称b7同源分子1(b7 homologue 1,b7-h1)是pd-1的配体,pd-l1与pd-1结合后,由pd-1传递抑制性信号,调控淋巴细胞的功能。此外,pd-1还可影响t细胞的分化,抑制炎性介质的释放,促进调节性t细胞(regulatory tcell,treg)的生成,从而在免疫应答的调控、免疫耐受的建立以及防止自身免疫性疾病中发挥重要作用。近几年发现,多种肿瘤细胞表面高表达pd-l1,通过与肿瘤浸润淋巴细胞表面的pd-1分子结合,抑制淋巴细胞的功能,是导致肿瘤发生免疫逃逸的重要原因之一。
[0007]
在肿瘤微环境中,pd-l1在肿瘤免疫逃逸中起了十分重要的作用。一方面,cd8 t细胞可被诱导产生pd-1分子,与肿瘤细胞表面的pd-l1结合后,抑制cd8 t细胞产生颗粒酶和穿孔素,另一方面,pd-l1也可表达在肿瘤相关巨噬细胞表面,它们与cd8 t细胞表面pd-1结合,也可诱导cd8 t细胞发生凋亡。此外,pd-l1还可促进肿瘤细胞发生上皮间质化而促进肿瘤的转移和浸润,在肿瘤的免疫逃逸中也起到了重要作用。
[0008]
当t细胞表面的pd-1与肿瘤细胞或肿瘤相关巨噬细胞表面的pd-l1相互作用时,这种相互作用会引起一系列信号传导应答,从而导致t淋巴细胞增殖和相关细胞因子的分泌受到抑制、肿瘤抗原特异性t细胞的凋亡和/或无法免疫,最终抑制免疫应答并促进肿瘤细胞的逃逸。靶向pd-1或pd-l1的单克隆抗体可通过特异性阻断pd-1/pd-l1的相互作用来破坏肿瘤的免疫耐受性,恢复肿瘤特异性t细胞对肿瘤细胞的杀伤功能,并实现肿瘤清除。
[0009]
靶向于pd-1/pd-l1的单抗药物最早于2014年问世,默克(merck)的pembrolizumab和百时美施贵宝(bristol-myers squibb)的nivolumab最早于2014年9月在美国上市,用于治疗第四期黑色素瘤,后陆续开发多种适应症。2016年3月,罗氏旗下基因泰克(genentech)研发的atezolizumab被美国fda批准治疗转移性膀胱癌,同年10月获批用于治疗肺癌。默克(merck serono sa)和辉瑞(pfizer inc)的avelumab最早于2017年3月获批上市,用于治疗默克尔细胞癌。2018年9月,fda批准赛诺菲和再生元制药公司研发的cemiplimab上市,用于治疗转移性皮肤鳞状细胞癌。赛尔基因(celgene corp)公司和阿斯利康的子公司(medimmune llc)研发的durvalumab最早于2017年5月获批上市,用于治疗转移性膀胱癌和转移性非小细胞肺癌。最新上市的是创新生物制剂(innovent biologicsinc)与礼来公司(eli lilly&co)合作研发的sintilimab以及上海君实生物医药科技有限公司的toripalimab,2019年2月首先在中国上市。
[0010]
许多癌症患者受益于针对pd-1/pd-l1的单克隆抗体。但是,研究发现pd-1/pd-l1抗体并非对所有癌症患者都是有效的。临床试验数据表明,单独的pd-1/pd-l1抗体的有效应答率为约20%。
[0011]
提高癌症免疫疗法中的有效应答率,特别是在对针对pd-1/pd-l1的单克隆抗体没有应答的患者的情况下,是当前肿瘤免疫治疗亟待解决的一个问题。
[0012]
与单克隆相比,小分子抑制剂除了具有较低的制造成本,更高的稳定性以及更好的组织和肿瘤渗透性外,还可以提供更好的治疗指数,不仅可以根据最佳药效参数更灵活地进行临床给药和口服给药,而且可以维持合理的半衰期和血药浓度以避免全身性免疫原性。因此,小分子抑制剂可单独或与治疗性抗体联合用药以提供有希望的替代治疗策略,以解决单抗药物的耐药性和低临床应答问题。
[0013]
wo2018006795和wo2019128918公开了新型靶向pd-1和pd-l1相互作用的小分子抑制剂。其中公开的小分子抑制剂在小鼠肿瘤模型中表现出抗肿瘤效果。
[0014]
到目前为止,尽管一些专利和出版物已经公开了一系列针对pd-1/pd-l1途径的小分子抑制剂,但尚无任何的小分子抑制剂获得批准上市。目前,靶向活性较好、作用机理比较明确的一类小分子抑制剂由百时美施贵宝所报导,但是,并没有提供进一步的体内活性表征,包括这些小分子的功效和安全性。为了满足国内市场的需求,迫切需要更多具有新颖骨架的替代化合物用于未来的临床应用。
技术实现要素:
[0015]
发明人通过对哌嗪二酮衍生物的研究,偶然发现一种哌嗪二酮衍生物具有显著的pd-1/pd-l1结合抑制活性。
[0016]
首先,本发明提供一种具有pd-1/pd-l1抑制活性的小分子化合物(化合物i),化学名:1-(3-(7-氟-1,4-二氧代八氢吡咯并[1,2-a]吡嗪-3-基)丙基)胍,分子量:271.3,化学式:c
11h18
fn5o2,化学结构式如式(1)所示:
[0017][0018]
所述化合物i还可以是其旋光异构体。
[0019]
所述化合物i还可以是其药用盐。
[0020]
所述化合物i的药用盐是在化合物结构的伯胺基部分形成酸式盐。
[0021]
所述药用盐选自无机酸和有机酸。
[0022]
所述无机酸为盐酸、氢溴酸、硫酸和磷酸中的任意一种。
[0023]
所述有机酸为醋酸、丙酸、丁酸、草酸、丙二酸、琥珀酸、己二酸、苯甲酸、苯丙酸、肉桂酸、硬脂酸、三氟醋酸、马来酸、富马酸、烟酸、苹果酸、柠檬酸、乳酸、羟基丁酸、乳糖酸、酒石酸、扁桃酸、葡萄糖酸、水杨酸、葡萄糖醛酸和抗坏血酸中的任意一种。
[0024]
其次,提供化合物i在制备肿瘤免疫治疗药物中的应用。
[0025]
所述肿瘤免疫治疗,其特征在于抑制肿瘤细胞pd-1/pd-l1结合。
[0026]
所述小分子抑制剂在pd-1/pd-l1结合试验中(例如wo2018006795所述的试验)的ic50小于0.5nm。
[0027]
所述肿瘤免疫治疗,其特征在于,所述肿瘤选自骨癌、胰腺癌、皮肤癌、头或颈癌、皮肤或眼内恶性黑色素瘤、子宫癌、卵巢癌、直肠癌、肛门区域癌、胃癌、睾丸癌、子宫癌、输卵管癌、子宫内膜癌、宫颈癌、阴道癌、外阴癌、霍奇金病、非霍奇金淋巴瘤、食道癌、小肠癌、内分泌系统癌、甲状腺癌、甲状旁腺癌、肾上腺癌、软组织肉瘤、尿道癌、阴茎癌、慢性或急性白血病(包括急性髓性白血病、慢性髓性白血病、急性淋巴细胞性白血病、慢性淋巴细胞性白血病)、儿童实体瘤、膀胱癌、肾癌、肾盂癌、中枢神经系统(cns)肿瘤、原发性cns淋巴瘤、肿瘤血管生成、脊髓轴肿瘤、脑干神经胶质瘤、垂体腺瘤、卡波西氏肉瘤、表皮样癌、鳞状细胞癌、t细胞淋巴瘤、环境诱导的癌症(包括由石棉诱导的肺癌)、不可切除或转移性黑色素瘤、转移性非小细胞肺癌、晚期肾细胞癌、复发性或进展性经典霍奇金淋巴瘤、复发性或转移性头颈部鳞状细胞癌、局部晚期或转移性尿道上皮癌、晚期肝细胞癌、转移性小细胞肺癌、msi-h/dmmr转移性结直肠癌、原发性纵隔大b细胞淋巴瘤、胃或胃食管交界处腺癌、肝细胞癌、默克尔细胞癌、具有至少10mmhg的间质液压(ifp)的癌症或这些癌症的组合。
[0028]
所述肿瘤优选为不可切除或转移性黑色素瘤、转移性非小细胞肺癌、晚期肾细胞癌、复发性或进展性经典霍奇金淋巴瘤、复发性或转移性头颈部鳞状细胞癌、局部晚期或转移性尿道上皮癌、晚期肝细胞癌、转移性小细胞肺癌、msi-h/dmmr转移性结直肠癌、原发性
纵隔大b细胞淋巴瘤、胃或胃食管交界处腺癌、宫颈癌、肝细胞癌或默克尔细胞癌。
[0029]
所述肿瘤进一步优选为宫颈癌、肾细胞癌、黑色素瘤、乳腺癌、结直肠癌或头颈鳞状细胞癌(hnscc)。
[0030]
所述肿瘤最优选为乳腺癌。
[0031]
所述肿瘤最优选为黑色素瘤。
[0032]
所述肿瘤最优选为结直肠癌。
[0033]
再次,本发明提供化合物i的制备方法,该方法包括:
[0034]
1)
[0035][0036]
将中间体i,中间体ii和edc.hci溶于dcm中,0℃下,缓慢滴入dipea后,室温反应。反应完全后,加入dcm稀释反应液,分别用氯化铵溶液和饱和食盐水洗涤,无水na2so4干燥,浓缩后经柱层析分离纯化,得到固体。
[0037]
2)
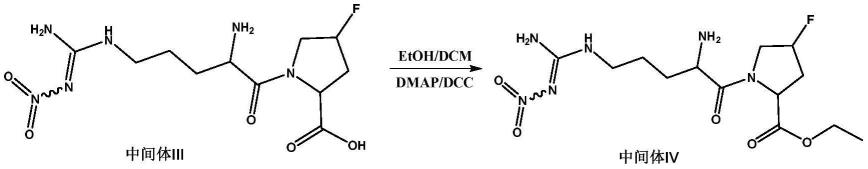
[0038][0039]
将中间体iii,乙醇,dcc和dmap的混合物在无水dcm中、在n2保护下于室温搅拌过夜。然后加入tea以及dcc和dmap,在n2保护下于室温搅拌过夜。然后加入dcc和dmap并且在n2保护下于室温搅拌2天。将混合物过滤并且将滤液用h2o洗涤四次,用na2so4干燥,过滤,并且在真空中除去溶剂。将产物在硅胶柱上纯化。然后将获得的白色固体在沸腾的乙酸乙酯中加热并且趁热过滤,得到中间体iv。
[0040]
3)
[0041][0042]
向中间体iv甲醇溶液中加入tea,然后将混合物在室温搅拌并蒸发。将残余物吸收在乙酸乙酯中,用hcl、饱和nahco3和盐水洗涤,na2so4干燥,并蒸发溶剂,得到中间体v,为白色固体。
[0043]
4)
[0044][0045]
将中间体v溶解在5%甲酸-甲醇中。将反应混合物在h2和pd/c下,在室温,0.2mpa下搅拌。过滤除去pd/c,蒸发反应混合物。获得白色粉末状的化合物i。
[0046]
最后,本技术提供包含化合物i的药物组合物。
[0047]
所述包含化合物i的药物组合物包括常规的药学上可接受的载体、赋形剂或稀释剂。“治疗有效量”可以根据受试者的类别、年龄、性别、严重程度和疾病类型、药物活性、对药物的敏感性、给药时间、给药途径、排泄率等来确定。药物组合物中的化合物i的量可以没有特别限制地广泛地变化,并且可以具体地为,相对于组合物的总量,0.00001重量%至50重量%,例如0.0001重量%至10重量%,或0.001重量%至5重量%,或0.1重量%至1重量%。
[0048]
可以将药物组合物配制成用于口服或非口服给药的固体、液体、凝胶或混悬剂形式,例如片剂、丸剂、散剂、颗粒剂、胶囊剂、乳剂、混悬剂、糖浆剂、乳化剂、浓缩液、灭菌水溶液、非水溶液、冻干制剂、栓剂等。
[0049]
对于口服施用,包含pd-1/pd-l1结合小分子抑制剂的药物组合物可以是片剂、胶囊、颗粒剂、液体胶囊、悬浮液或液体的形式。药物组合物优选地制成含有特定量的活性成分的剂量单位形式。例如,药物组合物可以以包含约0.1mg至1000mg范围内的化合物i的量的片剂或胶囊。
[0050]
所述药物组合物可以通过任何可接受和合适的口服制剂口服递送。口服制剂包括但不限于片剂、糖锭、锭剂、水性和油性悬浮液、可分散的粉剂或颗粒剂、乳剂、硬胶囊和软胶囊、液体胶囊、糖浆和酏剂。用于口服施用的药物组合物可以根据本领域已知的用于制造口服药物的任何方法来制备。为了提供药学上可接受的制剂,根据本公开的药物组合物可以包含至少一种选自以下的试剂:甜味剂、调味剂、着色剂、缓和剂、抗氧化剂和防腐剂。
[0051]
本发明的药物组合物,其口服给药的制剂含有常用的赋形剂,诸如粘合剂、填充剂、稀释剂、润滑剂、崩解剂、着色剂、助流剂、矫味剂和湿润剂,必要时可对片剂进行包衣。
[0052]
适用的填充剂包括纤维素衍生物、淀粉及淀粉衍生物、甘露糖醇、乳糖和其它类似的填充剂。
[0053]
适宜的崩解剂包括干淀粉、交联聚乙烯吡咯烷酮、淀粉衍生物,例如羧甲淀粉钠,交联羧甲基纤维素钠。
[0054]
适宜的润滑剂包括,例如硬脂酸镁,硬脂酰富马酸钠以及其他硬脂酸盐。
[0055]
适宜的药物可接受的润湿剂包括十二烷基硫酸钠。
[0056]
适宜的助流剂包括无水胶态二氧化硅、滑石粉等。
[0057]
适宜的粘合剂包括聚乙烯吡咯烷酮,羟丙基纤维素等粘性聚合物。
[0058]
可通过混合、制粒(干法制粒或湿法制粒)、干燥、填充,压片,包衣等常用的方法制备固体口服组合物。
[0059]
通过如下试验进一步说明本技术的有益效果:
[0060]
试验一、化合物i口服的急性毒性实验
[0061]
试验动物:取昆明种小白鼠80只,体重约为20g,预试验20只,正式试验60只,每组20只雌雄各半,灌胃给药。
[0062]
预试验:将化合物i加入蒸馏水,搅拌制备成最大浓度为0.4g/ml混悬液。参照霍恩(horn)氏方法,取小鼠20只,雌雄各半,随机分成4组,每组5只,分别灌胃给予不同剂量的化合物i,0.1ml/10g,0.2ml/10g,0.3ml/10g,0.4ml/10g,连续观察72小时,未见明显毒性反应。提示灌胃给药难以测出其ld
50
。
[0063]
最大给药试验:取小鼠60只,雌雄各半,随机分成3组,每组20只,分别灌胃给予化合物i样品混悬液,连续观察14天。给药剂量每次按0.2ml/10g,0.3ml/10g,0.4ml/10g给药,一日给药三次,连续给药14天。
[0064]
观察指标:给药后立即观察动物活动情况包括(小鼠的呼吸、自主活动与行为活动、眼检指征、唾液分泌、竖毛、肌张力、粪便、尿液等)及死亡情况。每天观察上述指标。
[0065]
试验结果:化合物i灌胃给药后,小鼠无明显中毒反应,呼吸正常,自主活动与行为活动无改变,未见眼球分泌物、眼球突出现象,粪便、尿液无异常。
[0066]
观察期间,试验动物摄食正常,体重无明显变化,情况良好。试验结束,离断颈椎处死小鼠进行解剖观察,心、肝、脾、肺、肾、睾丸、子宫等脏器未见明显病理改变。
[0067]
试验结果提示,在最大给药剂量0.4ml/10g,即按小鼠体重(20g),每次给予0.8g,每日三次,连续给予14天,对于试验动物是安全的。
[0068]
试验二、化合物i的体外抗肿瘤活性测定(参照中国专利cn 111793077 a施行)
[0069]
体外酶学水平的检测方法采用cisbio公司pd-1/pd-l1 binding assay kit检测试剂盒
[0070]
1.实验原理:
[0071]
pd-1蛋白带his标签,pd-1的配体pd-l1带hfc标签,分别用eu标记的anti-hfc抗体和xl665标记的anti-his抗体与两个标签蛋白结合。激光激发后,能量能够从供体eu上转移到受体xl665,使得xl665发光。而加入抑制剂后,阻断了pd-1与配体pd-l1的结合,使得eu和xl665距离较远,能量不能转移,xl665不发光。
[0072]
2.实验方法:
[0073]
使用cisbio bioassays的htrf测定试剂盒(目录号64icp01peg)进行pd-1/pd-l1结合测定,具体操作方法参照试剂盒说明进行。
[0074]
简单叙述如下,使用白色酶标板,将化合物i溶于dmso溶液中,浓度为1mm。首先将化合物i储备溶液用稀释剂稀释400倍,使用含有2.5%dmso的试剂盒稀释缓冲液稀释50倍,最后使用含有2.5%dmso的试剂盒稀释缓冲液依次稀释不同倍数。在每个孔中加入4μl的稀释液或稀释液稀释的化合物i。pd-1和pd-l1溶液分别以每孔3μl加入,化合物i与pd-1和pd-l1预孵育10分钟后,将按照产品说明书制备的10μl检测抗体加入到每个孔中。在室温下孵育平板过夜后,通过在pherastar fs酶标仪(bmg,德国)中读取平板获得数据。htrf信号计算为10000
×
(665/620比率)。将不同浓度化合物i的dmso溶液对计算信号拟合成具有可变斜率的s形剂量-反应曲线,并且通过曲线拟合(graphpad prism 7)获得ic 50值。
[0075]
实验结果显示,化合物i的ic50值为0.15nm。可见,本发明所述化合物i在分子水平
可显著抑制pd-1与pd-l1结合的相互作用,因此可用于治疗与pd-1/pd-l1的相互作用相关的疾病,尤其是肿瘤和自身免疫性疾病。
[0076]
对于本文中英文缩写的解释:
[0077]
ctla-4:细胞毒性t淋巴细胞相关蛋白-4
[0078]
pd-l1:细胞程序性死亡配体-1
[0079]
pd-1:细胞程序性死亡受体-1
[0080]
til:肿瘤浸润淋巴细胞
[0081]
itsm:immuno-receptor tyrosinebased switch motif,免疫受体酪氨酸转换基序
[0082]
shp-2:sh2 domain-containing protein-tyrosine phosphatase-2,蛋白酪氨酸磷酸酶-2
[0083]
his标签:多组氨酸标签
[0084]
dcm:二氯甲烷
[0085]
diepa:n,n-二异丙基乙基胺
[0086]
edc.hcl:1-乙基-(3-二甲基氨基丙基)碳化二亚胺盐酸盐
[0087]
dcc:二环己基碳二亚胺
[0088]
dmap:4-(n,n-二甲基氨基)吡啶
[0089]
n2:氮气
[0090]
h2:氢气
[0091]
meoh:甲醇
[0092]
etoh:乙醇
[0093]
tea:三乙胺
[0094]
pd/c:钯碳
[0095]
pbs:磷酸盐缓冲液
具体实施方式
[0096]
以下结合具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
[0097]
实施例1化合物i的合成
[0098]
1)
[0099][0100]
将中间体i(6.58g,30mmol),中间体ii(2.66g,20mmol)和edc.hci(5.78g,30mmol溶于dcm(100ml)中,0℃下,缓慢滴入dipea(21.0ml)后,室温反应3.0小时。反应完全后,加入dcm(200ml)稀释反应液,分别用氯化铵溶液和饱和食盐水洗涤,无水na2so4干燥,浓缩后经柱层析分离纯化(洗脱剂:石油醚/乙酸乙酯(v/v)=1/1)得到固体5.35g,产率:80%。
[0101]1h nmr(500mhz,chloroform-d)δ6.24(t,j=4.5hz,2h),5.08(tt,j=5.1,3.6hz,1h),4.98(tt,j=5.1,3.6hz,1h),4.83(d,j=6.5hz,4h),4.53(td,j=6.2,3.1hz,2h),3.84
–
3.76(m,5h),3.73
–
3.58(m,4h),3.27(qd,j=4.5,4.0,0.9hz,4h),2.28
–
2.11(m,5h),1.76
–
1.61(m,8h)。
[0102]
m/z:334.14。
[0103]
2)
[0104][0105]
将6.69g(20mmol)中间体iii,1.84g(40mmol)乙醇,8.25g(40mmol)dcc和3.67g(30mmol)dmap的混合物在150ml无水dcm中、在n2保护下于室温搅拌过夜。然后加入tea(5.4ml)以及7.5gdcc和3.4gdmap,在n2保护下于室温搅拌过夜。然后加入1gdcc和1.5gdmap并且在n2保护下于室温搅拌2天。将混合物过滤并且将滤液用h2o洗涤四次,用na2so4干燥,过滤,并且在真空中除去溶剂。将产物在硅胶柱(2-8%meoh-dcm)上纯化。然后将获得的白色固体在沸腾的乙酸乙酯中加热并且热过滤,得到6.16g中间体iv。
[0106]1h nmr(500mhz,chloroform-d)δ6.24(t,j=4.5hz,1h),5.08(tt,j=4.9,3.5hz,0h),4.97(tt,j=4.9,3.5hz,0h),4.83(d,j=6.5hz,2h),4.53(td,j=6.5,3.1hz,1h),4.13(q,j=6.3hz,2h),3.84
–
3.75(m,2h),3.72
–
3.57(m,2h),3.27(qd,j=4.5,4.0,0.9hz,2h),2.31
–
2.14(m,2h),1.76
–
1.61(m,4h),1.26(t,j=6.3hz,3h)。
[0107]
m/z:362.17
[0108]
3)
[0109][0110]
向中间体iv(7.25g,20mmol)在甲醇(100ml)中的溶液中加入7mltea,然后将混合物在室温搅拌18小时并蒸发。将残余物吸收在乙酸乙酯中,用1n hcl、饱和nahco3和盐水洗涤,干燥(na2so4)并蒸发溶剂,得到中间体v(5.38g,87%),为白色固体。
[0111]1hnmr(500mhz,chloroform-d)δ7.25(d,j=8.1hz,2h),6.30(t,j=4.5hz,2h),5.07(tt,j=5.1,3.7hz,1h),4.97(tt,j=5.1,3.7hz,1h),4.59(td,j=5.2,3.2hz,2h),4.46(dt,j=8.1,5.7hz,2h),3.82(s,3h),3.80
–
3.65(m,3h),3.29(qd,j=4.7,1.1hz,4h),2.33
–
2.16(m,4h),1.76
–
1.69(m,1h),1.71
–
1.66(m,1h),1.69
–
1.61(m,2h),1.65
–
1.58(m,2h),1.61
–
1.53(m,2h).
[0112]
m/z:316.13。
[0113]
4)
[0114][0115]
将中间体v(6.33g,20mmol)溶解在5%甲酸-甲醇(100ml)中。将反应混合物在h2和10%pd/c下在室温,0.2mpa下搅拌4小时。过滤除去pd/c,蒸发反应混合物。获得白色粉末状的式(i)化合物5.43g。
[0116]1h nmr(500mhz,chloroform-d)δ7.61(t,j=3.6hz,1h),7.25(d,j=8.1hz,1h),6.78(s,1h),6.26(s,1h),5.07-4.97(td,j=5.1,3.7hz,1h),4.59(td,j=5.2,3.2hz,1h),4.46(dt,j=8.1,5.8hz,1h),3.81
–
3.65(m,2h),3.07(qd,j=4.9,3.6hz,2h),2.33
–
2.16(m,2h),1.76
–
1.51(m,4h).
[0117]
m/z:271.14。
[0118]
实施例2化合物i在b16-f10模型中的抗肿瘤功效的体内试验(参照中国专利cn112587666a施行)
[0119]
实验所需的材料:
[0120]
实验动物:40只c57bl/6小鼠,雌性,约6-8周龄,约17-21g,分为空白对照组,低剂量组,中剂量组和高剂量组,每组10只。
[0121]
配制材料:
[0122]
dmem培养基;
[0123]
胎牛血清;
[0124]
甲基纤维素(mc)。
[0125]
细胞制备和植入:在37℃下,在5%co2的空气气氛中,将b16-f10肿瘤细胞在补充有10%热灭活的胎牛血清的dmem培养基中作为单层培养物体外保存。通过胰蛋白酶-edta处理,每周三次常规传代培养肿瘤细胞。收获生长到约70%-80%汇合的细胞,并计数用于肿瘤接种。
[0126]
肿瘤细胞接种和分组施用:将含有1x106b16-f10肿瘤细胞(悬浮在基础dmem培养基中的细胞)的100μl细胞悬浮液接种到小鼠的右侧皮下。接种后第二天,根据肿瘤接种的顺序,采用分层随机进行分组,并在分组当天开始给药。
[0127]
测试物质的制备:
[0128]
化合物i溶液的制备:称量化合物i约0.1g,在搅拌条件下加入到100ml纯化水中,溶解,得澄清透明的水溶液。作为供试样品,4℃冷藏,备用。
[0129]
给药:
[0130]
将空白对照组中的小鼠称重并记录。对空白对照组中的小鼠根据其体重通过口服施用每天两次给予纯化水(即空白对照),容量为0.1ml/10g。
[0131]
将低剂量组中的小鼠称重并记录。对该组中的小鼠根据其体重通过口服施用每天两次给予所制备的化合物i水溶液,容量为0.1ml/10g。
[0132]
将中剂量组中的小鼠称重并记录。对该组中的小鼠根据其体重通过口服施用每天
两次给予所制备的化合物i水溶液,容量为0.2ml/10g。
[0133]
将高剂量组中的小鼠称重并记录。对该组中的小鼠根据其体重通过口服施用每天两次给予所制备的化合物i水溶液,容量为0.4ml/10g。
[0134]
每周用数字游标卡尺测量肿瘤三次并计算肿瘤体积。如果肿瘤的大小超过2000mm3,或动物具有严重的疾病、疼痛或不能自由进食和饮水,则实施安乐死。每天通过电子天平测量动物的体重。当动物明显瘦弱并且其体重减少超过20%时,则需要安乐死。在施用化合物i溶液后20天,实验结束。
[0135]
肿瘤抑制率计算:
[0136]
tgi(%)=(1-(给药当天的肿瘤体积-给药第一天的肿瘤体积)/(给药当天的肿瘤体积-媒介物组第一天的肿瘤体积)x100%。
[0137]
使用graphpad prism 7.0软件,通过双向方差分析来分析小鼠的肿瘤体积变化,并根据邦费罗尼事后检验方法与空白对照组比较,p《0.05被认为是显著不同的。
[0138]
结果表明,化合物i可以显著抑制小鼠皮下移植的黑色素瘤细胞系的生长,并且其在c57bl/6小鼠中具有良好的耐受性,且没有明显的不良反应。结果总结在表2中。
[0139]
表2:化合物i在b16-f10荷瘤小鼠模型中的抗肿瘤功效的体内试验结果
[0140][0141][0142]
上述实验结果显示,化合物i口服后对于b16-f10荷瘤小鼠模型呈现显著的肿瘤抑制作用。与空白对照组相比,高中低三个剂量组均能显著抑制肿瘤的增长,并且随着给药剂量的增加,肿瘤抑制率明显增加。说明口服化合物i对于b16-f10荷瘤小鼠具有优良的肿瘤抑制活性,可以用于进一步开发成抗肿瘤药物。
[0143]
以上发明内容和实施例描述了本发明专利申请的基本原理和主要特征及本发明专利申请优点,本领域的技术人员应该了解,本发明专利申请不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明专利申请的最优的技术方案,在不脱离本发明专利申请精神和范围的前提下,本发明专利申请还会有各种变化和改进,即含有化合物i的任何口服给药剂型及其处方组成,都落入要求保护的本发明专利申请范围内,本发明专利申请要求保护范围由所附的权利要求书及其等效物界定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
