一种基于reads特征评估肿瘤来源的方法与流程
- 国知局
- 2024-07-12 10:35:48
本发明属于肿瘤细胞识别,具体来说,涉及一种基于reads特征评估肿瘤来源的方法。
背景技术:
1、人体降解的dna片段通过细胞凋亡、坏死或细胞主动分泌释放到血液中,可作为细胞游离dna(cfdna)进行检测。循环肿瘤dna(ctdna)是指实体肿瘤细胞内的dna经脱落或者当细胞凋亡后释放进入循环系统的游离dna片段,是一种特征性的肿瘤生物标记。临床研究表明,ctdna浓度与肿瘤负荷成正比。cfdna的半衰期低于1个小时,通过ctdna的检测可实时有效监测肿瘤进展。已有研究证明ctdna检测相较于影像学能够提前1个月到半年提示肿瘤的复发。采用基于ctdna的液体活检可有效检测肿瘤复发,并在新辅助治疗疗效预测,辅助治疗疗效预测,免疫治疗疗效监测,晚期患者的系统治疗疗效监测,药物假期提示等方面具有探索,并表现出良好的应用前景。
2、mrd是指实体肿瘤经过根治性后依旧残留在体内不可见的分子残留病灶(molecular residue disease),这些病灶通过影像学无法检测,但依然导致肿瘤复发或转移。ngs(二代测序技术)可以快速,高通量的检测患者体内残留的微量或不可见的肿瘤循环dna(ctdna),检测类型包括突变检测,甲基化检测,片段组学检测。其中突变检测可以检测单碱基替换,缺失和插入等,不仅可以提示患者的mrd状态,并且可为患者监控肿瘤进展的新发突变。
3、但是在mrd肿瘤负荷较低的临床环境中,ctdna仅占cfdna的一小部分,通常小于0.1%。目前ngs测序方法的错误率在万分之一左右,因此在ctdna应用中准确区分错误与真实突变是非常重要的。为了增加mrd检测的敏感性,可以通过增加测序深度提高mrd的敏感性,但是随着测序深度的增加,会检出更多的假阳性位点。为了提高mrd检测的敏感性和特异性,需要通过实验技术和生信算法改进mrd检测性能。
4、集成数字错误抑制(ides)是华盛顿大学医学院aaron m newman等人发明的一种基于肿瘤不知情(tumor-agnostic assays)策略进行癌症个性化分析的方法,用于检测ctdna。该方法结合分子标签技术和基于健康人背景噪音的抛光(polish)策略,在非小细胞肺癌(non-small cell lung cancer,nsclc)患者水平达到92%的敏感性和>99%的特异性。但是该方法需要较多的健康人构建背景噪音数据库,增加了策略的应用成本,并且只可以应用于肿瘤不知情策略,不适用固定化面板(fixed-panel)以外的个性化追踪位点,无法准确的判断变异是否为真实的体细胞突变。分子残留病灶检测(mrdetect)技术是纽约基因组中心asafzviran等人实现的通过全基因组测序(wgs)定量cfdna肿瘤负荷,该算法提取追踪位点的突变reads特征,包括变异碱基质量,read平均质量、read方向、read正负链、突变位置和比对质量,使用支持向量机(svm)机器学习方法抑制测序错误,该方法未使用突变背景,片段信息,分子标签信息等特征,无法在单个突变水平定性mrd状态。
技术实现思路
1、本发明提供了一种基于reads特征评估肿瘤来源的方法,抑制pcr扩增和测序过程中产生的背景错误,发现cfdna中真实的肿瘤突变。
2、为实现上述技术目的,本发明采用的技术方案如下:
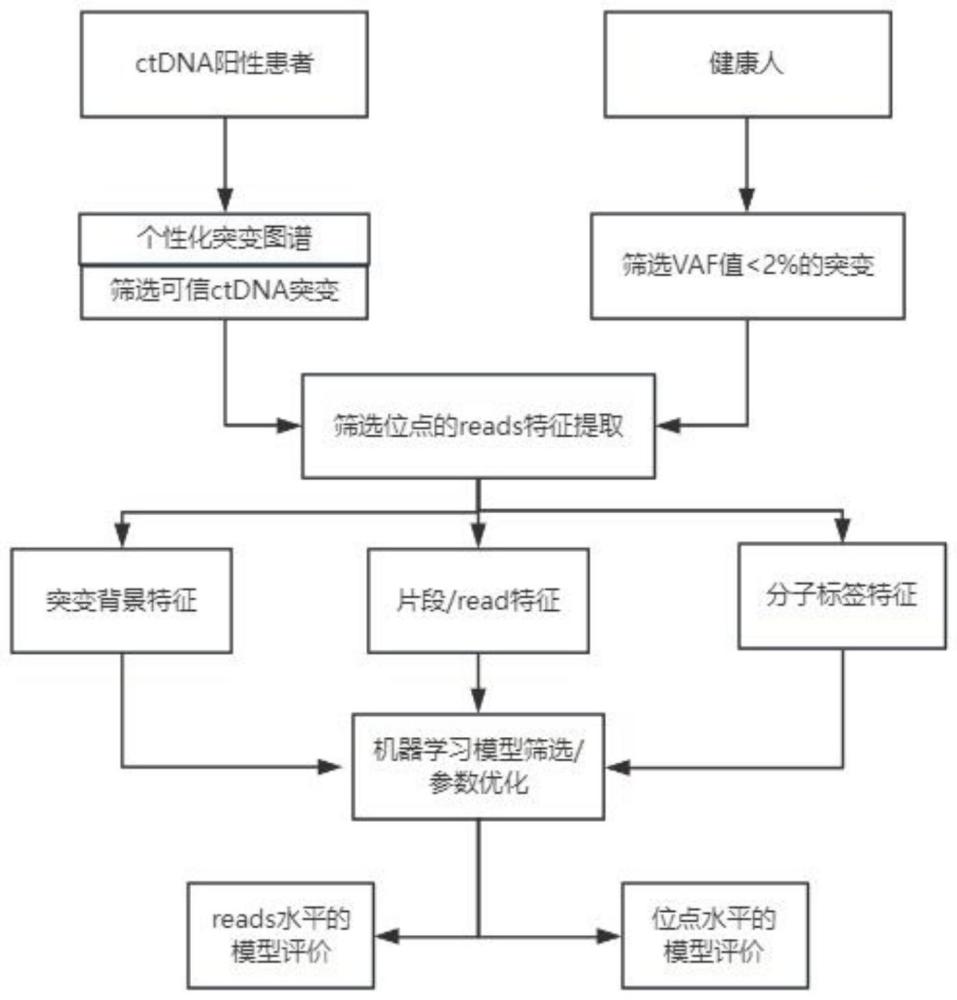
3、一种基于reads特征评估肿瘤来源的方法,包括步骤:
4、s1、阳性ctdna突变集筛选和阴性cfdna突变集筛选;
5、s2、根据基因组fasta文件和比对bam文件进行特征提取;
6、s3、将阳性集和阴性集reads做好肿瘤与非肿瘤标签,将数据集随机分成n份;
7、s4、基于多个机器学习模型算法构建分类模型,并通过5折交叉验证方法进行模型准确性及稳定性评估,筛选最优模型;
8、s5、利用网格搜索方法找到模型最优参数组合,并进行模型性能测试;
9、s6、将reads回溯至位点水平,单个位点的肿瘤概率为该位点所有reads肿瘤概率的平均值,进行模型性能测试。
10、进一步地,阳性ctdna突变集筛选包括:ctdna突变和背景噪音及其他突变;从cfdna突变图谱中筛选ctdna突变,挑选位点深度≥500,突变深度≥3,vaf(变异等位基因分数)>0.003,等突变位点,共计筛选500个ctdna突变位点,共计约5000条reads作为阳性集。
11、进一步地,阴性突变集筛选:去除胚系突变后,vaf(变异等位基因分数)<0.02,位点深度≥500,从10例健康人筛选出30000条reads;随机挑选5000条reads作为阴性集。
12、进一步地,步骤s2中所提取的特征包括:
13、单碱基突变类型(g>a):单碱基突变;
14、三碱基突变类型(tg>ac):单碱基突变,包括突变两侧的碱基;
15、突变位置的基因组背景:突变两侧5bp参考碱基和突变碱基的基因组序列占比;insert size:read1和read2之间插入片段的长度;
16、read:突变发生在read1或read2;
17、orient:突变比对到基因组正链或负链;
18、position:突变位置距离read 5’端的距离;
19、duplex read:是否为dcs(双端一致性序列)reads;
20、base质量:包括突变碱基质量、突变两侧2bp、3bp、5bp、整体read的平均碱基质量;
21、ad,cd:分子标签去重矫正前的reads数量;
22、ae,ce:分子标签去重矫正前的碱基错误率。
23、进一步地,测序数据获得:
24、肿瘤患者组织样本及白细胞对照处理:
25、组织样本和对照样本基因组dna进行超声打碎,建成gdna文库;将dna文库分别与内部1123panel(2m大小)进行杂交,将基因组dna目标区域捕获下来;回收目标dna片段,构建高通量测序文库。
26、肿瘤患者和健康人cfdna样本处理:
27、根据组织生信分析结果生成患者个性化突变图谱,合成个性化探针,加入内部200kpanel探针,共同用于cfdna捕获(健康人只使用200k panel);将以上探针与cfdna进行杂交;回收目标cfdna,构建高通量测序文库。
28、进一步地,生信处理:
29、肿瘤患者组织样本及白细胞对照处理:
30、获取fastq文件:在高通量测序仪(mgi2000)上完成测序,测序平台将得到的光信号转化为bcl格式的测序下机数据,并对下机数据进行拆分,根据样本index将单个样本的测序数据拆分出来,转换成fastq格式。
31、获取高质量bam文件:将第一步获取的fastq文件进行数据质控,通过数据质控去除测序低质量的序列;利用基因组bwa比对软件进行比对,获取bam文件,使用samtools去除冗余,得到去冗余的bam文件;使用samtools过滤mapq值低于30的序列生成高质量的去冗余bam文件,之后使用gatk对bam文件进行矫正,获得矫正之后的bam文件。
32、获取肿瘤变异检测vcf文件和注释文件:通过变异检测软件gatk,检测肿瘤突变,得到vcf文件,使用anovar,snpeff软件对突变位点注释,注释内容包括,突变类型,人群频率,cosmic数据库记录。
33、获取患者个性化肿瘤突变集,筛选条件:去除胚系突变,位点质控合格,组织位点深度>50,突变深度>8,人群频率<0.002,非链特异性。
34、肿瘤患者和健康人cfdna样本处理:
35、获取fastq文件:在高通量测序仪(mgi2000)上完成测序,测序平台将得到的光信号转化为bcl格式的测序下机数据,并对下机数据进行拆分,根据样本index将单个样本的测序数据拆分出来,转换成fastq格式。
36、获取高质量bam文件:将第一步获取的fastq文件进行数据质控,通过数据质控去除测序低质量的序列;利用fgbio软件提取umi序列,并添加至umi.ubam文件;将umi.ubam文件转为fastq文件,利用基因组bwa比对软件进行比对,获得umi.bam文件;使用picard软件,合并umi.ubam和umi.bam文件,或者umi.merge.bam文件;通过fgbio软件进行umi矫正,对umi序列一致的reads进行去重矫正,获得双端(dcs)和单端一致性序列(scs)。
37、获取肿瘤变异检测vcf文件和注释文件:通过变异检测软件gatk,检测肿瘤突变,得到vcf文件,使用anovar,snpeff软件对突变位点注释,注释内容包括,突变类型,人群频率,cosmic数据库记录。
38、本发明相比现有技术,具有如下有益效果:
39、突变背景特征,reads特征的,片段特征的选择;提取特征来源数据包括但不限于panel,wes,wgs等二代测序方法;该算法适用于tumor-informed策略和策略;ctdna突变集来源于自有的突变图谱数据库。降低背景噪音的构建成本,该方法有很好的鲁棒性。
本文地址:https://www.jishuxx.com/zhuanli/20240614/87924.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表