基于二代测序的基因融合分析方法、产品及应用与流程
- 国知局
- 2024-07-12 10:37:40
本发明涉及基因检测领域,具体涉及基于二代测序的基因融合分析方法及系统。本发明更具体涉及基于二代测序技术对dna水平的全基因组测序或靶向测序数据进行分析,准确地检测目的基因的融合变异。本发明的方法灵敏度高、计算资源消耗小,且能够减少假阳性信号产生。
背景技术:
1、在肿瘤患者的基因组中,会大量携带多种体细胞变异,包括点突变、拷贝数变异以及结构性变异等。其中,基因融合是一种与肿瘤的发生和发展密切相关的结构性变异。基因融合在基因组中是普遍存在的,通常是由于染色体重排等因素,两个不相关的基因发生了染色体易位、中间缺失或染色体倒置形成一个新基因的过程,其对原有基因功能产生了影响。融合基因的发现始于20世纪60年代,在慢性粒性白血病病人中发现的费城染色体,拉开了融合基因与疾病的研究序幕。
2、诸多研究表明,基因融合与众多疾病,特别是癌症的发生发展密切相关,甚至是一些癌症的直接诱因。许多疾病的发生都伴随有融合基因现象,例如,白血病常伴随有bcr-abl、aml1-eto、cbfβ-myh11、pml-rarα等融合基因;在多种实体瘤中也发现融合基因,如非小细胞肺癌中的eml4-alk,前列腺癌中的slc45a3-elk4,横纹肌肉瘤中的pax3-foxo1等等。科学研究发现,一些融合基因参与了相关疾病的致病过程。因此,融合基因的检测可作为诊断标准之一,也可能作为治疗靶点,或用于药物筛选,从而成为当前组学大数据分析中的一项重要研究内容。
3、在当前的肿瘤精准诊疗过程中,基因融合作为肿瘤的驱动突变或特定的药物治疗靶点,其准确检测在临床辅助诊断和靶向治疗方面均具有重要意义。例如,在非小细胞肺癌中,基因融合检测已成为肺癌常规诊疗中的一部分。
4、在临床应用过程中,可以分别通过诸如ihc法、fish法、rt-pcr法和二代测序方法对基因融合进行检测。其中,ihc方法虽然耗时短,成本低,但当缺乏优秀的特异性结合抗体时,其灵敏度和特异性表现均较差;fish方法通过分离探针的设计可以同时检测已知融合和未知融合,但对于某些罕见或未知的断裂与融合位点间距较小时则难以检测,同时其检测过程比较复杂,对操作和判读技术要求较高,不适合用于大规模的筛查性检测;rt-pcr方法周期短、敏感性高、操作简单,但其主要局限性为只能基于rna序列检测已知的基因融合类型,不能检测未知的融合类型。基于dna序列的二代测序方法,可以同时对已知融合和未知融合进行检测。对于石蜡包埋组织(ffpe)或血浆游离核酸等特殊样本类型,使用dna进行测序其成功率和适用范围均相较于rna更高。因此,基于dna序列进行二代测序的方法,是基因融合检测的重要技术手段。
5、基于dna序列的二代测序需要对所有目的基因的内含子、外显子进行同时测序,得到的数据量较大,对于下游的数据分析软件提出了更高的算法要求。目前,常见的基因融合分析方法有三种:
6、1、基于已知融合位置构建参考序列,对测序序列逐一进行比对。此方法理论检出能力较高,但计算量巨大,且无法对未知融合进行检测,使用范围较小。
7、2、分析比对到不同染色体上或比对距离显著超出文库插入片段范围的双端序列,进而查找基因融合事件。此方法检测速度快,资源消耗少,但检出能力稍弱。
8、3、对测序序列进行不同长度的迭代打断,对两侧序列进行二次序列比对,对于同一个序列比对到两个位置的信号进行收集,进行融合检测。此方法检测灵敏度较高,但运算量较大,对计算资源的需求较高。
9、同时,基于dna测序的基因融合检测需要分析大量的内含子区域序列,这些序列的保守性较低,会出现大量错误拼接,导致最终的输出结果包含大量假阳性信号,影响临床判断。
10、cn108073791a公开了一种基于二代测序数据检测目标基因结构变异的方法,其从测序序列的两个前端开始进行循环比对来进行基因结构变异的检测。但该技术不对所得序列进行过滤,检测特异性差。
11、cn114005490a公开了一种基于二代测序技术的循环肿瘤dna融合检测方法,其中,通过采用序列-数字转换方法比较序列的相似性,来提高软件的运行速度,降低软件对资源的需求,但并非从测序序列的两个前端进行循环比对,算力需求仍然较大。
12、cn114743594a公开了一种用于结构变异检测的方法、装置和存储介质,其中包括数据获取步骤、信号分类步骤、dp信号聚类分析步骤、融合断点分析步骤、sr信号分析步骤、计算和注释步骤和注释结果合并和输出步骤,但该技术注重于识别包括缺失、倒位、重复、染色体内易位、染色体间易位等多种结构变异类型,不是专用于基因融合的检测。
13、综上所述,本领域中急需开发出灵敏度高、计算资源消耗小,且能够减少假阳性信号产生的基因融合分析方法和系统。
技术实现思路
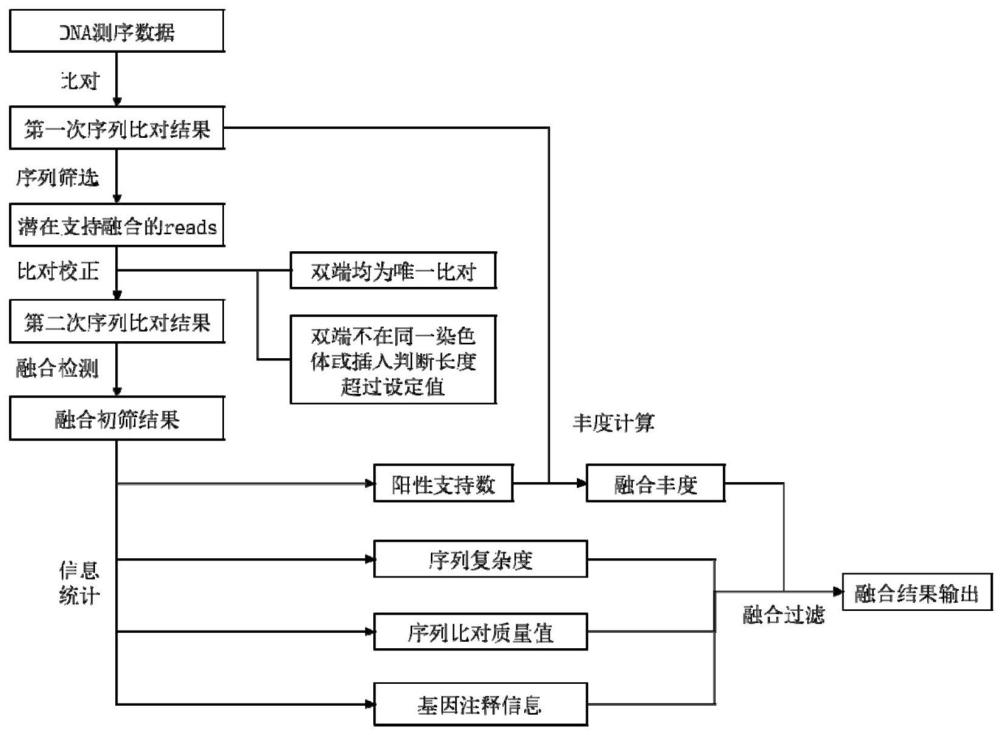
1、本发明人使用二代测序数据,对基因融合的分析方法进行研究。本发明人认为如果一个dna片段支持融合事件,则序列插入片段两端的测序起始位置必然跨越融合断点,因此从序列两端向中间进行逐次比对校准,能够以最小的计算量,获得最佳的基因融合检测效果。基于上述理论,发明人开发了一种灵敏度高、计算资源消耗小,且能够减少假阳性信号产生的基因融合分析方法。本发明适用于基于二代测序的全基因组测序或靶向区域测序。
2、基于上述发现,本发明的一个方面涉及一种基因融合分析方法,其包括:
3、输入二代测序数据,
4、经过融合初筛过程,获得阳性支持序列。
5、在一些实施方式中,本发明的方法还包括在融合初筛过程后经过融合过滤过程,对阳性支持序列进行过滤,将最终保留的阳性支持序列作为具有融合阳性信号的序列。
6、在一些实施方式中,融合初筛过程包括以下步骤:
7、基于二代测序数据进行第一次序列比对;
8、根据第一次序列比对结果,对序列进行第一次序列筛选;
9、从reads1和reads2的起点开始,对序列进行循环迭代的全基因组比对;
10、对循环迭代的全基因组比对后的数据进行融合检测,获得阳性支持序列。
11、在一些实施方式中,融合过滤过程包括通过选自以下的一个或多个方式,对阳性支持序列进行过滤:
12、分别对阳性支持序列中上游基因的序列比对质量值和下游基因的序列比对质量值进行统计,获得比对质量值;
13、对序列上存在的单碱基或多碱基的重复度进行计算,获得序列复杂度;
14、对阳性支持序列进行计数,获得阳性支持数;
15、对野生型和突变型的比对深度或序列条数进行统计,获得融合丰度值;
16、根据融合的比对位置,查询相应的基因信息,获得融合基因的位置注释。
17、本发明的另一个方面涉及一种基于二代测序的基因融合分析系统,其特征在于,包括输入模块、融合初筛模块和输出模块,所述输入模块用于导入二代测序数据的输入单元;所述融合初筛模块包括第一次序列比对单元、第一次序列筛选单元、二次比对校正单元、融合检测单元;所述输出模块用于输出具有融合阳性信号的序列。
18、在一些实施方式中,本发明的系统还包括融合过滤模块,所述融合过滤模块包括选自下组的一个或多个比对:质量值计算单元、序列复杂度计算单元、阳性支持数计算单元、融合丰度计算单元、融合基因的位置注释单元。
19、本发明的又一个方面涉及一种基于二代测序的基因融合分析装置,其中,所述装置包括存储器和处理器;存储器包括用于存储实现本发明的基于二代测序的基因融合分析方法的程序;处理器包括用于通过执行存储器存储的程序以实现本发明的基于二代测序的基因融合分析方法。
20、本发明的再一个方面涉及一种计算机可读存储介质,其中,所述存储介质中存储有程序,该程序能够被处理器执行以实现本发明的基于二代测序的基因融合分析方法。
21、本发明的再一个方面涉及本发明的基因融合分析系统、本发明的基因融合分析装置或如本发明的计算机可读存储介质在制备用于基因融合相关疾病的诊断、基因融合相关疾病预后评估、基因融合相关疾病用药指导、基因融合相关药物靶点筛选的产品中的应用。
22、采用本发明,则可在灵敏度高、计算资源消耗小,且能够减少假阳性信号产生的情况下分析获得序列中存在基因融合的序列。
本文地址:https://www.jishuxx.com/zhuanli/20240614/88144.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表