一种镥[177Lu]氧奥曲肽前体化合物的制备方法与流程
- 国知局
- 2025-01-10 13:43:26
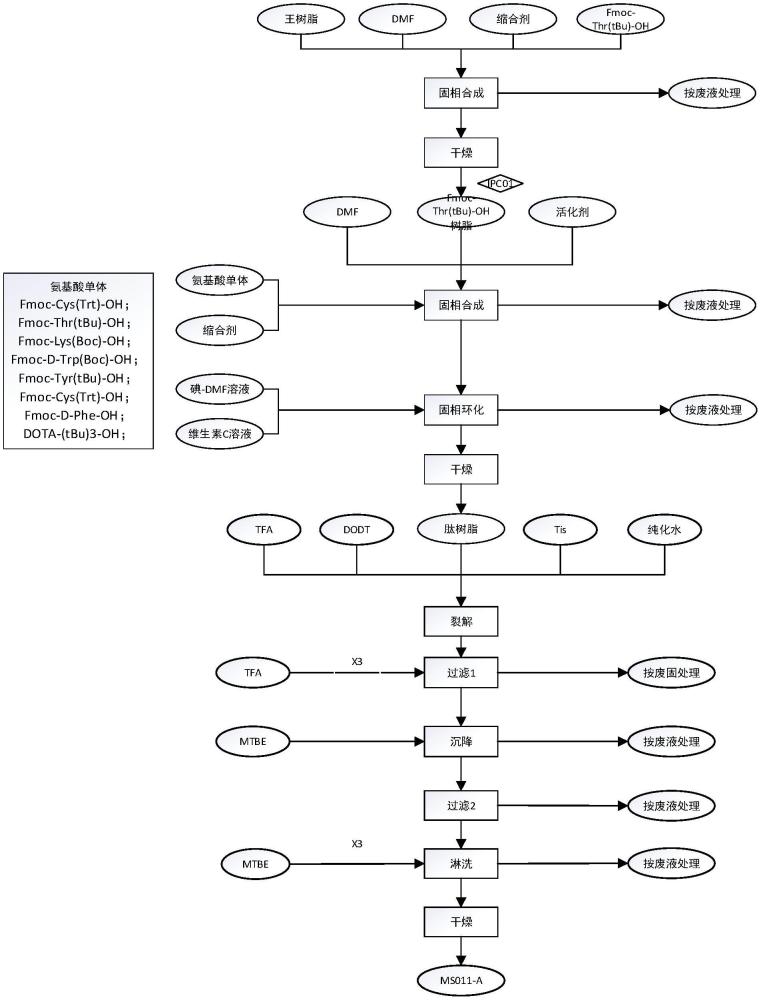
本发明属于制药。具体地,本发明涉及一种镥[177lu]氧奥曲肽前体化合物的制备方法及其应用。
背景技术:
1、2018年,美国fda批准了诺华子公司法国advanced accelerator applications公司的镥[177lu]氧奥曲肽(lutathera)上市,将其用于治疗标准治疗后进展的sstr阳性胃肠胰腺神经内分泌肿瘤(gep-net)。镥[177lu]氧奥曲肽作为第一个获得批准的用于放射性配体治疗(rlt)的放射性药物,其通过与肿瘤细胞表面生长激素抑制素受体的特异性结合,到达肿瘤细胞并释放核素产生辐射以杀伤肿瘤细胞,而且近期的临床试验结果还表明其临床应范围还有希望进一步扩大。
2、镥[177lu]氧奥曲肽是通过核素[177lu]与作为螯合剂的以下式i所示的前体化合物(ms011)螯合而制成的,该前体化合物由8个氨基酸组成的多肽连接螯合剂dota(1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸,1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)构成,其中十二烷杂环上的四个氮原子加上与氮原子连接的四个乙酸基团所形成的八齿配体提供电子,从而与位于中心的镥[177lu]原子形成稳定的螯合体。
3、放射性核素偶联药物的工业化生产对于前体化合物制备方法的收率和产物纯度都提出了很高的要求,杂质会严重影响后续核素标记的过程。而现有的镥[177lu]氧奥曲肽前体化合物制备方法还普遍存在着收率不高、纯度偏低的问题,这对于实现镥[177lu]氧奥曲肽原料药的高效规模化生产以满足临床用药需求提出了重大挑战。因此,改进镥[177lu]氧奥曲肽前体化合物的制备工艺成为制药领域中亟待解决的技术问题之一。
技术实现思路
1、鉴于此,本发明目的在于提供一种镥[177lu]氧奥曲肽前体化合物的制备方法,该制备方法能够显著提升所制备的镥[177lu]氧奥曲肽前体化合物的纯度,降低杂质含量,提高产物收率,简化后续纯化工艺,为镥[177lu]氧奥曲肽的规模化工业化生产和临床推广应用奠定了基础。
2、本发明的上述目的是通过以下技术方案实现的:
3、一种式i所示的镥[177lu]氧奥曲肽前体化合物的制备方法,其包括通过固相合成法,使fmoc-thr(tbu)-oh、fmoc-cys(trt)-oh、fmoc-thr(tbu)-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh、fmoc-cys(trt)-oh、fmoc-d-phe-oh和dota-(tbu)3-co2h在王树脂上依次进行偶联反应,得到dota-(tbu)3-d-phe-cys(trt)-tyr(tbu)-d-trp(boc)-lys(boc)-thr(tbu)-cys(trt)-thr(tbu)-王树脂,再进行氧化反应使其中的两个cys脱除trt保护基并且彼此之间形成二硫键从而环化,然后进行裂解反应去除王树脂和保护基,得到式i所示的镥[177lu]氧奥曲肽前体化合物,
4、
5、根据本发明的一些实施方案,所述制备方法包括以下步骤:
6、(1)在包含缩合反应试剂的有机溶剂中,使fmoc-thr(tbu)-oh在王树脂上进行偶联反应,得到fmoc-thr(tbu)-王树脂;
7、(2)在包含缩合反应试剂的有机溶剂中,使fmoc-cys(trt)-oh、fmoc-thr(tbu)-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh、fmoc-cys(trt)-oh和fmoc-d-phe-oh在fmoc-thr(tbu)-王树脂上依次进行偶联反应,得到fmoc-d-phe-cys(trt)-tyr(tbu)-d-trp(boc)-lys(boc)-thr(tbu)-cys(trt)-thr(tbu)-王树脂;
8、(3)在包含缩合反应试剂的有机溶剂中,使dota-(tbu)3-co2h在fmoc-d-phe-cys(trt)-tyr(tbu)-d-trp(boc)-lys(boc)-thr(tbu)-cys(trt)-thr(tbu)-王树脂上进行偶联反应,得到dota-(tbu)3-d-phe-cys(trt)-tyr(tbu)-d-trp(boc)-lys(boc)-thr(tbu)-cys(trt)-thr(tbu)-王树脂;和
9、(4)在包含氧化剂的有机溶剂中进行氧化反应,使得dota-(tbu)3-d-phe-cys(trt)-tyr(tbu)-d-trp(boc)-lys(boc)-thr(tbu)-cys(trt)-thr(tbu)-王树脂中的两个lys脱去trt保护基并彼此之间形成二硫键从而环化;
10、(5)在包含三氟乙酸(tfa)、2,2’-(1,2-乙二基双氧代)双乙硫醇(dodt)、三异丙基硅烷(tis)和纯化水的混合液中进行裂解反应去除王树脂和保护基,得到式i所示的镥[177lu]氧奥曲肽前体化合物,其中所述裂解反应在5~10℃下进行1小时,然后在26~30℃下进行3小时。
11、根据本发明的一些实施方案,在步骤(1)中,在进行所述偶联反应之前在有机溶剂中溶胀王树脂。
12、根据本发明的一些实施方案,在步骤(1)中,所述偶联反应在25~30℃下进行2小时;优选地,所述fmoc-thr(tbu)-oh先在0~10℃,优选5℃下活化3~5分钟,再在25~30℃下进行偶联反应2小时。
13、根据本发明的一些实施方案,在步骤(1)中,所述缩合反应试剂包含以下组合之一:六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(pybop)、1-羟基苯并三唑(hobt)和n,n-二异丙基乙胺(diea);o-苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐(hbtu)、1-羟基苯并三唑、n,n-二异丙基乙胺和4-二甲氨基吡啶(dmap);或者1-羟基苯并三唑、n,n'-二异丙基碳二亚胺(dic)和4-二甲氨基吡啶。优选地,所述缩合反应试剂包含六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、1-羟基苯并三唑和n,n-二异丙基乙胺。其中,所述fmoc-thr(tbu)-oh、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、1-羟基苯并三唑和n,n-二异丙基乙胺的摩尔当量比可以为1:1:1:2;所述fmoc-thr(tbu)-oh、o-苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐、1-羟基苯并三唑、n,n-二异丙基乙胺和4-二甲氨基吡啶的摩尔当量比可以为1:1:1:2:0.1;所述fmoc-thr(tbu)-oh、1-羟基苯并三唑、n,n'-二异丙基碳二亚胺和4-二甲氨基吡啶的摩尔当量比可以为1:1:1:0.1。
14、根据本发明的一些实施方案,在步骤(1)中,所述fmoc-thr(tbu)-王树脂的取代度为0.40~0.70mmol/g。
15、根据本发明的一些实施方案,在步骤(1)中,还包括在包含封端剂和有机碱的有机溶剂中,对得到的fmoc-thr(tbu)-王树脂进行封端;优选地,所述封端剂为乙酸酐(ac2o);其中,所述有机碱可以为n,n-二异丙基乙胺和/或三乙胺,优选为n,n-二异丙基乙胺;优选地,所述封端在20~30℃下进行15~20小时。
16、根据本发明的一些实施方案,在步骤(2)中,所述偶联反应在18-32℃,优选20~30℃下进行2~3小时;优选地,所述fmoc-cys(trt)-oh、fmoc-thr(tbu)-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh、fmoc-cys(trt)-oh或fmoc-d-phe-oh先在0~10℃,优选5℃下活化5~10分钟,再在18~32℃,优选20~30℃下进行偶联反应2~3小时。
17、根据本发明的一些实施方案,在步骤(2)中,所述缩合反应试剂包含以下组合之一:1-羟基苯并三唑和n,n'-二异丙基碳二亚胺;o-苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐、1-羟基苯并三唑和n,n-二异丙基乙胺;或者六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷和n,n'-二异丙基碳二亚胺。优选地,所述缩合反应试剂包含:1-羟基苯并三唑和n,n'-二异丙基碳二亚胺。其中,所述fmoc-cys(trt)-oh、fmoc-thr(tbu)-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh、fmoc-cys(trt)-oh或fmoc-d-phe-oh、1-羟基苯并三唑和n,n'-二异丙基碳二亚胺摩尔当量比可以为1:1.05:1.05;所述fmoc-cys(trt)-oh、fmoc-thr(tbu)-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh、fmoc-cys(trt)-oh或fmoc-d-phe-oh、o-苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐、1-羟基苯并三唑和n,n-二异丙基乙胺的摩尔当量比可以为1:1:1:2;所述fmoc-cys(trt)-oh、fmoc-thr(tbu)-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh、fmoc-cys(trt)-oh或fmoc-d-phe-oh、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷和n,n'-二异丙基碳二亚胺的摩尔当量比可以为1:1:2。
18、根据本发明的一些实施方案,在步骤(2)中,所述fmoc-cys(trt)-oh、fmoc-thr(tbu)-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh、fmoc-cys(trt)-oh或fmoc-d-phe-oh与fmoc-thr(tbu)-王树脂的摩尔当量之比独立地为2~4:1,优选为3:1。
19、根据本发明的一些实施方案,在步骤(2)中,还包括在采用kaiser检测所述偶联反应产物为阴性后延长反应0.5~2小时。
20、根据本发明的一些实施方案,在步骤(2)中,还包括进行所述偶联反应之前,在包含脱保护剂的有机溶剂中分别脱除fmoc-thr(tbu)-oh、fmoc-cys(trt)-oh、fmoc-thr(tbu)-oh、fmoc-lys(boc)-oh、fmoc-d-trp(boc)-oh、fmoc-tyr(tbu)-oh和fmoc-cys(trt)-oh引入的fmoc保护基的步骤;其中,所述脱保护剂可以为哌啶(pip)、或哌啶和1,8-二氮杂双环[5.4.0-7]十一碳-7-烯,优选为哌啶;优选地,所述脱除fmoc保护基的步骤在20~30℃下进行两次,每次15~30分钟,优选20分钟。
21、根据本发明的一些实施方案,在步骤(3)中,所述偶联反应在20~30℃下进行2~3小时;优选地,所述dota-(tbu)3-co2h先在0~10℃下活化5~10分钟,再在20~30℃下进行偶联反应2~3小时。
22、根据本发明的一些实施方案,在步骤(3)中,所述缩合反应试剂包含以下组合之一:2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、1-羟基-7-偶氮苯并三氮唑和n,n-二异丙基乙胺;o-苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐、1-羟基苯并三唑和n,n-二异丙基乙胺;或者六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、1-羟基苯并三唑和n,n-二异丙基乙胺。优选地,所述缩合反应试剂包含六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、1-羟基苯并三唑和n,n-二异丙基乙胺。其中,所述dota-(tbu)3-co2h、2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯、1-羟基-7-偶氮苯并三氮唑和n,n-二异丙基乙胺的摩尔当量比可以为1:0.98:1:2;所述dota-(tbu)3-co2h、o-苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐、1-羟基苯并三唑和n,n-二异丙基乙胺的摩尔当量比可以为1:0.98:1:2;所述dota-(tbu)3-co2h、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、1-羟基苯并三唑和n,n-二异丙基乙胺的摩尔当量比可以为1:1:1:2。
23、根据本发明的一些实施方案,在步骤(3)中,还包括进行所述偶联反应之前,在包含脱保护剂的有机溶剂中脱除fmoc-d-phe-oh引入的fmoc保护基的步骤;其中,所述脱保护剂可以为哌啶(pip)、或哌啶和1,8-二氮杂双环[5.4.0-7]十一碳-7-烯,优选为哌啶;优选地,所述脱除fmoc保护基的步骤在20~30℃下进行两次,每次15~30分钟,优选20分钟。
24、根据本发明的一些实施方案,在步骤(4)中,所述氧化反应在27~32℃下进行25~55分钟,优选为35~45分钟。
25、根据本发明的一些实施方案,在步骤(4)中,所述氧化剂为碘;优选地,碘与fmoc-thr(tbu)-王树脂的摩尔当量之比为3~5:1,优选为4:1。优选地,碘在反应体系中的浓度为0.15~0.25mol/l,优选为0.2mol/l。
26、根据本发明的一些实施方案,在步骤(4)中,还包括在所述氧化反应之后,在包含还原剂的有机溶剂中,对得到的环化的dota-(tbu)3-d-phe-cys-tyr(tbu)-d-trp(boc)-lys(boc)-thr(tbu)-cys-thr(tbu)-王树脂进行洗涤;其中,所述还原剂可以为维生素c和/或亚硫酸钠,优选为维生素c;优选地,所述洗涤在20~30℃下进行两次,每次10分钟。
27、根据本发明的一些实施方案,在步骤(5)中,所述三氟乙酸、2,2’-(1,2-乙二基双氧代)双乙硫醇、三异丙基硅烷和纯化水的体积比为80~90:4~10:3~5:3~5,优选为90:4:3:3。
28、根据本发明的一些实施方案,在步骤(5)中,所述包含三氟乙酸(tfa)、2,2’-(1,2-乙二基双氧代)双乙硫醇(dodt)、三异丙基硅烷(tis)和纯化水的混合液与步骤(4)得到的环化的树脂的用量比为10~30ml:1g,优选为30ml:1g。
29、根据本发明的一些实施方案,在步骤(5)中,还包括将所述裂解反应得到的产物过滤,并将滤液于≤20℃的温度下在甲基叔丁基醚(mtbe)中静置沉降15~20小时。优选地,甲基叔丁基醚与包含三氟乙酸(tfa)、2,2’-(1,2-乙二基双氧代)双乙硫醇(dodt)、三异丙基硅烷(tis)和纯化水的混合液的体积比为10~12:1。
30、根据本发明的一些优选实施方案,在步骤(1)~(5)中,所述有机溶剂独立地为n,n-二甲基甲酰胺(dmf)和/或n-甲基-2-吡咯烷酮,优选为n,n-二甲基甲酰胺。
31、此外,本发明还提供了一种镥[177lu]氧奥曲肽的制备方法,其包括将根据本发明的式i所示的镥[177lu]氧奥曲肽前体化合物的制备方法得到的式i所示的镥[177lu]氧奥曲肽前体化合物与镥[177lu]螯合。
32、本发明至少具有如下有益效果:
33、本发明提供了优化的镥[177lu]氧奥曲肽前体化合物的制备方法,其降低了前体化合物粗品中单个杂质含量并提高了前体化合物产物纯度,使得产物纯度≥90.0%,更有利于后续的纯化工序的进行,从而实现单个杂质含量≤0.10%重量,为镥[177lu]氧奥曲肽的规模化工业生产和临床应用奠定了基础。
本文地址:https://www.jishuxx.com/zhuanli/20250110/354937.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。