一种基于多组学数据分析的泛癌分析方法及系统
- 国知局
- 2024-07-11 17:33:22
本发明涉及一种基于多组学数据分析的泛癌分析方法及系统,属于生物信息。
背景技术:
1、泛癌:泛癌(pan-cancer)是指多种不同类型的癌症共享一些共同的生物学特征,如基因突变、信号通路异常等,这些特征可能是癌症发生和发展的共性机制,为癌症的早期诊断和治疗提供了新的思路。
2、转录组:转录组是由dna转录生成的rna的集合,包括编码蛋白的信使rna(messenger rna,mrna)和各种非编码rna,如mirna、lncrna等。
3、dna甲基化:dna甲基化是一种很重要的表观遗传修饰事件,是指在不改变基因组dna序列的前提下,将甲基基团可逆地添加到dna的胞嘧啶上。dna甲基化的状态与癌症密切相关,异常的甲基化能够在早期癌症中被检测到。
4、癌症诊断:癌症诊断指的是通过血液检测、组织样本检测、成像检测等多技术手段确定癌症的类型和所处阶段进行判断和预测。及时、准确的癌症诊断,对于制定治疗方案、提高治愈率和改善患者生活质量是非常关键的。
5、癌症诊断模型的目标是找到一些生物分子特征作为诊断标志物,并基于这些标志物构建预测模型。中国专利申请号cn202211280689.9采用图卷积神经网络处理转录组学数据实现肺癌的早期诊断。但是,由于癌症的异质性,不同类型的癌症需要提取不同的生物标志物,这使得癌症的早期诊断工作更加复杂且成本更高。传统的癌症诊断模型也仅能够对已知类型的癌症进行建模诊断,难以识别新型癌症的发生。而且,癌症的发生时多组学数据共同作用的结果,transformer结构可以增强模型的上下文理解能力,从而为癌症组学数据提供更好的聚类效果。
技术实现思路
1、本发明的目的在于提供一种基于多组学数据分析的泛癌分析方法及系统,以解决如何使用多组学数据获得一种泛化能力强、准确率更高的泛癌分析方法。
2、一种基于多组学数据分析的泛癌分析方法,所述方法包括:
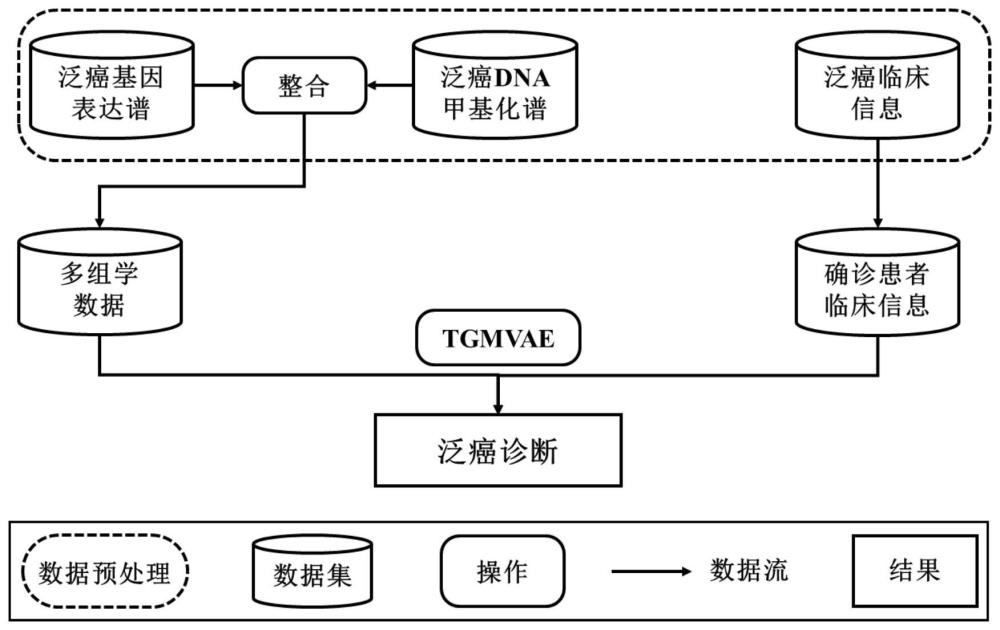
3、从tcga数据库中整合多种癌症的多组学数据,其中,多组学数据包括转录组和表观基因组数据及对应临床信息;
4、对多组学数据进行预处理,得到处理后的数据;
5、将处理后的数据输入预先训练的基于transformer的高斯混合变分自编码器聚类模型,输出分析结果,并对结果进行可视化解释和泛癌分析表构建。
6、进一步地,所述从tcga数据库中集中多种癌症的多组学数据的方法包括:
7、从nih网站进入tcga gdc数据库,下载带tcga标签的癌症的基因表达数据、dna甲基化数据和临床信息注释文件。
8、进一步地,所述基因表达数据由rna-seq技术测量得到,所述dna甲基化数据由methylation array技术测量得到,所述临床标注信息用于提取患者的确诊信息,并作为诊断结果验证标签使用。
9、进一步地,所述从tcga gdc数据库下载得到的癌症的基因表达数据包括:brca、lihc、lusc、coad、aml、kirc、ov、sarc、skcm。
10、进一步地,所述从tcga gdc数据库下载得到的癌症的dna甲基化数据包括:brca、lihc、lusc、coad、aml、kirc、ov、sarc、skcm。
11、进一步地,所述对多组学数据进行预处理的方法包括:
12、(a)根据临床信息区分出多中癌症患者中的癌症样本和正常样本,剔除正常样本,将癌症样本整理在一个矩阵中,行是样本,列是特征;
13、(b)数据过滤,对于样本的过滤,去除单一组学数据中含有大量特征缺失值的噪声样本,以及多组学数据中组学不完整的样本,其次,对特征进行过滤,过滤掉缺失值大于特征总量10%的特征以及表达水平过低或者表达水平变化幅度小的分子特征;
14、(c)缺失值处理,采用k近邻法进行缺失值填充,使用缺失值的10个近邻特征的加权值来补齐缺失数据,以最大化地保留数据的原有特征分布;
15、(d)数据标准化,对数据进行对数标准化,以消除数据的偏差和异方差性,其次,采用最大-最小标准化,将每个特征的数值缩放到0到1之间;
16、(e)将临床信息中的tcga编号作为样本唯一的索引值,保留与之对应的多组学数据,并合并其特征维度。
17、进一步地,所述基于transformer的高斯混合变分自编码器聚类模型的训练方法包括:构建初始基于transformer的高斯混合变分自编码器聚类模型;
18、采用训练数据集作为初始模型输入进行训练;
19、计算训练过程中基于transformer的高斯混合变分自编码器的神经网络拟合损失以及生成样本与输入样本之间产生的重构损失,选取其中模型损失loss最低的模型参数为预先训练的基于transformer的高斯混合变分自编码器聚类模型。
20、进一步地,所述基于transformer的高斯混合变分自编码器聚类模型的函数表达式为:
21、loss=lossrec+lossclu=crossentropy(x,x;)+dkl(p(z|k)||p(z,k|x))
22、其中,前项lossrec为基于transformer的变分自编码器重构样本模型损失,使用交叉熵计算输入样本x与输出新样本x’之间的误差;后项lossclu为基于高斯混合模型对样本进行聚类的聚类损失,使用kl散度计算样本的真实分布p(z,k|x)与模型拟合的潜在空间分布p(z|k)之间的聚类误差,z表示模型的潜在空间变量,k表示样本的聚类数。
23、进一步地,所述基于transformer的高斯混合变分自编码器聚类模型包括:
24、编码器,用于将输入的高维样本数据映射到一个低维的潜在空间中,使映射的样本集和在潜在空间中符合混合高斯分布,配合解码器使用,以实现新样本的生成功能;
25、解码器,用于在潜在空间的混合高斯分布中进行随机采样并通过神经网络拟合来重构样本,即生成新样本;
26、transformer模块,用于在编码器和解码器中各层mlp中进行重加权操作;
27、高斯混合模型,用于配合编码器和解码器构成的变分自编码器的数据生成过程,其由均值μ、方差σ2和高斯混合系数π线性组合而成,变分自编码器训练过程中进行模型拟合,高斯混合模型的输入是编码器映射的低维潜在空间样本,输出是潜在空间的混合高斯分布,配合解码器的采样和样本重构,同时,高斯混合模型通过样本相似性将输入样本划分为若干个不用的高斯分量,依次达到样本聚类效果,通过可视化分析高斯混合模型产生的聚类可以实现泛癌诊断分析;
28、主干网络,用于综合编码器潜在空间分布拟合过程中产生的拟合损失和解码器重构样本与输入样本之间的重构损失,以达到本分析系统的整体损失最小,误差最小。
29、一种基于多组学数据分析的泛癌分析系统,所述系统包括:
30、数据获取模块,从tcga数据库中整合多种癌症的多组学数据,其中,多组学数据包括转录组和表观基因组数据及对应临床信息;
31、数据处理模块,对多组学数据进行预处理,得到处理后的数据;
32、分析模块,将处理后的数据输入预先构建的基于transformer的高斯混合变分自编码器聚类模型,输出分析结果,并对结果进行可视化解释和泛癌分析表构建。
33、与现有技术相比,本发明所达到的有益效果:
34、1、本发明在特征学习网络中引入了transformer模块,帮助诊断模型提取组学数据内部依赖性特征,并且克服了长序列数据中的长距离遗忘的问题,增强了特征提取网络的学习能力,进而提高了分析模型性能;
35、2、该发明使用聚类算法进行泛癌诊断可以减少模型对标签数据的依赖并且保留模型对新癌症的发现能力,这是模型更具泛化能力。
本文地址:https://www.jishuxx.com/zhuanli/20240615/84875.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表