五配位单原子催化剂及其制备方法和应用
- 国知局
- 2024-11-21 11:39:31
本发明属于催化剂,具体来说涉及一种五配位单原子催化剂及其制备方法和应用。
背景技术:
1、亚砜类化合物在工业化社会中无处不在,因为在形成硫醚键后,碳氢化合物的物理和化学性质会得到广泛的修改。因此,亚砜类化学品广泛用于制药、农用化学品、精细化学品以及其他有价值的化学品的生产。尽管亚砜类化合物具有重要的实用性,但选择性地将硫醚氧化成亚砜仍是一项具有挑战性的任务,为此已经开发了各种氧化合成策略。传统上,使用无机氧化剂如三氧化铬、高碘酸和其他高价碘化物等能源密集型方法来合成亚砜。然而,这些过程伴随着大量或过量的氧化剂的使用,导致能源消耗大和温室气体排放,并且含有大量有毒副产品。此外,由于氧化反应无法控制,过氧化生成砜的问题也很常见。因此,迫切需要设计一种简单、快速、可控、环保且实用的方法来选择性合成亚砜类化合物。
2、单线态氧(1o2),一种具有温和氧化能力(2.2v vs nhe)和未占据π*轨道的活性氧物种(ros),在电子丰富的底物氧化中显示出高度的亲电性和选择性,对绿色有机合成发挥着重要作用。电催化和光催化技术可以在常温条件下将分子氧转化为1o2,用于硫醚的选择性合成,但由于氧气溶解度低和催化材料性质的限制,这些技术仍面临着效率低下、规模化生产难以及使用易腐蚀金属电极的挑战。最近,催化剂在类芬顿反应方面的进展为选择性生成1o2提供了重要机会。其中,基于过氧单硫酸盐(pms)的类芬顿反应为1o2生成提供一个几乎完美的路径。不幸的是,由于pms的固有电子受体和供体特性以及其与催化剂上多个活性位点的氧化还原反应,pms基类芬顿系统中的1o2生成通常伴随着其他ros(例如·oh和so4·-)的共存,这最终导致底物的过度氧化。此外,通过类芬顿系统介导的亚砜类化合物的合成很少被探索。为了进一步探索pms介导的1o2氧化过程的应用潜力,迫切需要在开发具有高活化效率和高选择性1o2生成的新催化剂方面取得突破。
3、特别值得关注的是,过渡金属单原子催化剂(sacs)因其强大的金属-载体相互作用和可定制的活性金属位点,在pms生成1o2领域受到广泛关注。li等人证实,锚定在n掺杂碳支撑上的co-n4基团可以作为1o2生成的主要活性位点,而xu等人的后续研究表明,co-n4位点负责生成自由基和非自由基。上述结果可能归因于金属原子细微的配位环境差异而显著影响其活性中心原子的电子结构,从而影响反应中间体的吸附能垒,进而影响反应途径和选择性。可见金属原子的配位环境十分重要。
技术实现思路
1、针对现有技术的不足,本发明的目的在于提供一种五配位单原子催化剂,所述五配位单原子催化剂为含有五配位co单原子的co-n5纳米催化剂,五配位co单原子是指有五个n原子与co原子配位。
2、本发明的另一目的在于提供上述五配位单原子催化剂的制备方法,该制备方法通过氯化铵引入n原子来构建五配位的co单原子,通过氯化铵的加入量来调控五配位单原子催化剂的性能,控制g-c3n4上co单原子位点的配位环境。
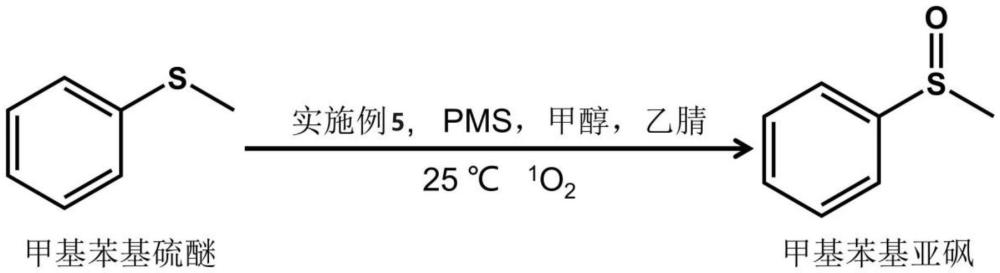
3、本发明的另一目的在于提供上述五配位单原子催化剂在合成甲基苯基亚砜中的应用,该五配位单原子催化剂能活化pms实现100%的1o2产生,从而解决目前催化剂活化pms产生1o2选择性低的问题。
4、本发明的目的是通过下述技术方案予以实现的。一种五配位单原子催化剂的制备方法,包括:将g-c3n4、钴盐和铵盐混合均匀,得到第一粉末,在惰性气体氛围下,将所述第一粉末于500~580℃保温4~5h,洗涤,干燥,研磨,得到五配位单原子催化剂,其中,所述g-c3n4的质量份数、钴盐中钴元素的物质的量份数和铵盐中铵根离子的物质的量份数的比为0.5:0.25:(4~12),所述质量份数的单位为g,物质的量份数的单位为mmol。
5、在上述技术方案中,所述g-c3n4的质量份数、钴盐中钴元素的物质的量份数和铵盐中铵根离子的物质的量份数的比优选为0.5:0.25:8,所述质量份数的单位为g,物质的量份数的单位为mmol。
6、在上述技术方案中,获得所述g-c3n4的方法为:将双氰胺于540~560℃保温4~5h,冷却至室温,得到g-c3n4。
7、在上述技术方案中,双氰胺于540~560℃保温前,将双氰胺研磨至少5min,优选5~10min。
8、在上述技术方案中,所述钴盐为乙酰丙酮钴。
9、在上述技术方案中,所述铵盐为氯化铵。
10、在上述技术方案中,升温至500~580℃的升温速率为2~4℃/min。
11、在上述技术方案中,升温至540~560℃的升温速率为2~4℃/min。
12、在上述技术方案中,所述洗涤采用水,洗涤用于除去可溶性钴盐。
13、在上述技术方案中,所述干燥的温度为60~80℃,干燥的时间为12~14h。
14、在上述技术方案中,将g-c3n4、钴盐和铵盐混合后研磨5~10min,以实现所述混合均匀。
15、上述五配位单原子催化剂在合成甲基苯基亚砜中的应用,五配位单原子催化剂活化pms使甲基苯基硫醚合成甲基苯基亚砜。
16、在上述技术方案中,将五配位单原子催化剂和pms放置于甲基苯基硫醚混合溶液中,在搅拌条件下进行类芬顿催化反应。
17、在上述技术方案中,所述甲基苯基硫醚混合溶液为甲基苯基硫醚和溶剂的混合物,所述溶剂为能够同时溶解甲基苯基硫醚、甲基苯基亚砜和pms的物质。
18、在上述技术方案中,所述甲基苯基硫醚混合溶液中甲基苯基硫醚的浓度为1.9~2.1mmol/l。
19、在上述技术方案中,按质量份数计,所述五配位单原子催化剂和pms的比为(5~10):8。
20、本发明首次实现了通过五配位单原子催化剂活化pms将甲基苯基硫醚定向转化为甲基苯基亚砜,五配位单原子催化剂活化pms能实现1o2的100%产生。相比于四配位co单原子催化剂,在50min时,甲基苯基亚砜合成的选择性从7.5%(四配位co单原子催化剂)提高到99.2%(五配位单原子催化剂),产率从10.2%(四配位co单原子催化剂)提高到99.6%(五配位单原子催化剂)。
21、与已有技术相比,本发明具有以下优点:
22、1.本发明制备的五配位单原子催化剂活化pms合成甲基苯基亚砜的过程中能实现1o2的100%产生。
23、2.本发明的五配位单原子催化剂能高效活化pms,甲基苯基亚砜的产率为99.6%。
24、3.本发明通过氯化铵调控合成的五配位单原子催化剂在活化pms将甲基苯基硫醚转化为甲基苯基亚砜过程中,对甲基苯基亚砜合成的选择性高达99.2%。
25、4.本发明所制备的co-n5/pms系统相较于电催化和光催化甲基苯基亚砜合成系统具有更优异的反应速率。
26、5.本发明所制备的五配位单原子催化剂具有高的稳定性,能够多次重复使用。
技术特征:1.一种五配位单原子催化剂的制备方法,其特征在于,包括:将g-c3n4、钴盐和铵盐混合均匀,得到第一粉末,在惰性气体氛围下,将所述第一粉末于500~580℃保温4~5h,洗涤,干燥,研磨,得到五配位单原子催化剂,其中,所述g-c3n4的质量份数、钴盐中钴元素的物质的量份数和铵盐中铵根离子的物质的量份数的比为0.5:0.25:(4~12),所述质量份数的单位为g,物质的量份数的单位为mmol。
2.根据权利要求1所述的制备方法,其特征在于,获得所述g-c3n4的方法为:将双氰胺于540~560℃保温4~5h,冷却至室温,得到g-c3n4。
3.根据权利要求2所述的制备方法,其特征在于,所述钴盐为乙酰丙酮钴;所述铵盐为氯化铵。
4.根据权利要求3所述的制备方法,其特征在于,将g-c3n4、钴盐和铵盐混合后研磨5~10min,以实现所述混合均匀。
5.如权利要求1~4所述制备方法获得的五配位单原子催化剂。
6.如权利要求5所述五配位单原子催化剂在合成甲基苯基亚砜中的应用,五配位单原子催化剂活化pms使甲基苯基硫醚合成甲基苯基亚砜。
7.根据权利要求6所述的应用,其特征在于,将五配位单原子催化剂和pms放置于甲基苯基硫醚混合溶液中,在搅拌条件下进行类芬顿催化反应。
8.根据权利要求7所述的应用,其特征在于,所述甲基苯基硫醚混合溶液为甲基苯基硫醚和溶剂的混合物,所述溶剂为能够同时溶解甲基苯基硫醚、甲基苯基亚砜和pms的物质。
9.根据权利要求8所述的应用,其特征在于,所述甲基苯基硫醚混合溶液中甲基苯基硫醚的浓度为1.9~2.1mmol/l。
10.根据权利要求6所述的应用,其特征在于,按质量份数计,所述五配位单原子催化剂和pms的比为(5~10):8。
技术总结本发明公开了一种五配位单原子催化剂及其制备方法和应用,所述制备方法包括:将g‑C<subgt;3</subgt;N<subgt;4</subgt;、钴盐和铵盐混合均匀,得到第一粉末,在惰性气体氛围下,将所述第一粉末于500~580℃保温4~5h,洗涤,干燥,研磨,得到五配位单原子催化剂。本发明首次实现了通过五配位单原子催化剂活化PMS将甲基苯基硫醚定向转化为甲基苯基亚砜,五配位单原子催化剂活化PMS能实现<supgt;1</supgt;O<subgt;2</subgt;的100%产生。相比于四配位Co单原子催化剂,在50min时,甲基苯基亚砜合成的选择性从7.5%提高到99.2%,产率从10.2%提高到99.6%。技术研发人员:展思辉,赵志勇,张涛,杨梦雪,杨高华,李轶,岳帅受保护的技术使用者:南开大学技术研发日:技术公布日:2024/11/18本文地址:https://www.jishuxx.com/zhuanli/20241120/332266.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表