用于在甲基化分区测定中分析无细胞dna的组合物和方法
1.相关申请的交叉引用
2.本技术要求2019年9月30日提交的美国临时专利申请第62/908,569号的优先权的权益,为了所有目的将该申请通过引用并入本文。
发明领域
3.本公开内容提供了涉及分析dna(诸如无细胞dna)的组合物和方法。在一些实施方案中,无细胞dna来自患有或怀疑患有癌症的受试者和/或无细胞dna包括来自癌细胞的dna。在一些实施方案中,将dna分区为第一子样品和第二子样品,其中第一子样品包含比第二子样品具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的dna,并且使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序,以及以区分第一子样品的dna中的第一核碱基和第二核碱基的方式对dna进行测序。
4.引言及概述
5.癌症每年导致全球数百万人死亡。因为早期癌症趋向于对治疗更易感,因此癌症的早期发现可以导致改进的结果。
6.不适当控制的细胞生长是癌症的标志,通常由以下遗传改变和表观遗传改变的积累导致:诸如拷贝数变异(cnv)、单核苷酸变异(snv)、基因融合、插入和/或缺失(插入/缺失(indel))、包括胞嘧啶的修饰(例如,5-甲基胞嘧啶、5-羟甲基胞嘧啶和其他更氧化的形式)以及dna与染色质蛋白和转录因子的缔合的表观遗传变异。
7.活检是用于检测或诊断癌症的常规方法,其中从癌症的可能部位提取细胞或组织,并分析相关的表型和/或基因型特征。活检具有侵入性的缺点。
8.基于体液(诸如血液)分析(“液体活检”)来检测癌症,是基于对从癌细胞释放到体液中的dna的观察的令人感兴趣的替代方法。液体活检是非侵入性的(有时仅需要抽血)。然而,由于无细胞dna的低浓度和异质性,开发用于分析提供关于核碱基修饰的详细信息的液体活检材料的准确且灵敏的方法一直具有挑战性。分离和处理可用于液体活检程序中进一步分析的无细胞dna的级分(fraction)是这些方法的重要部分。因此,需要用于分析无细胞dna(例如,在液体活检中)的改进的方法和组合物。
9.本公开内容部分地基于以下实现。与其他过程步骤(诸如基于甲基化程度分区和测序)串联(in-line)分析核碱基修饰(包括尤其是胞嘧啶的甲基化和/或羟甲基化)可以是有益的。例如,在示例性实施方案中,将dna样品(诸如cfdna样品)分区为多于一个具有不同胞嘧啶甲基化的量的子样品(例如,基于与mbd(甲基结合结构域或甲基结合蛋白)或对甲基化胞嘧啶特异性的抗体的结合),并且然后使包含高甲基化水平的子样品经历差异性影响特定核碱基的不同形式(例如,未修饰的胞嘧啶和甲基化胞嘧啶,或羟甲基化胞嘧啶和甲基化胞嘧啶)的程序。然后可以进行测序以鉴定第一子样品和第二子样品中的序列和/或鉴定来自第一子样品的dna中特定种类的核碱基存在的位置。根据本公开内容的这样的方法可以比现有方法(诸如medip-seq、mbd-seq、bs-seq、ox-bs-seq、tap-seq、ace-seq、hmc-seal和tab-seq)提供更多关于dna(诸如cfdna)中表观遗传修饰的信息。参见,例如,schutsky,
e.k.等人nondestructive,base-resolution sequencing of 5-hydroxymethylcytosine using a dna deaminase.nature biotech,2018;doi.10.1038/nbt.4204(ace-seq);yu,miao等人base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome.cell,2012;149(6):1368-80(tab-seq);han,d.a highly sensitive and robust method for genome-wide 5hmc profiling of rare cell populations.mol cell.2016;63(4):711-719(5hmc-seal);shen,s.y.等人sensitive tumour detection and classification using plasma cell-free dna methylomes.nature.2018;563(7732):579-583(cfmedip);nair,ss等人comparison of methyl-dna immunoprecipitation(medip)and methyl-cpg binding domain(mbd)protein capture for genome-wide dna.epigenetics.2011;6(1):34-44。与这样的现有方法不同,根据本公开内容的方法可以凭借分区步骤提供关于第一修饰(诸如甲基化水平)的组合信息和凭借差异性影响特定核碱基的不同形式的程序提供关于特定修饰和/或其位置的另外的信息。这样的程序的实例包括使用亚硫酸氢盐、取代硼烷、碱基修饰酶或区分一类核碱基的不同种类的修饰的碱基特异性抗体的各种转化或分离步骤。在一些实施方案中,本文描述的方法提供了关于以下信息的组合:(i)分子的总体修饰(例如,胞嘧啶修饰)水平(例如,基于分子的分区)和(ii)关于特定修饰的身份和/或位置的更高分辨率信息(例如,基于特定修饰或未修饰的核苷酸的特定转化或基于区分特定修饰类型的进一步分区,随后进行测序,如本文详细讨论的)。
10.本发明的方法还可以包括从dna中捕获两个靶区组。在一些实施方案中,靶区组包括序列可变靶区组和表观遗传靶区组。这些组中的每一个都可以提供对确定样品含有来自癌细胞的dna的可能性有用的信息。在一些实施方案中,序列可变靶区组的捕获产量大于表观遗传靶区组的捕获产量。捕获产量的差异可以允许在序列可变靶区组中深且因而更准确的序列确定,并允许在表观遗传靶区组中浅而宽的覆盖率,例如,在同时测序期间,诸如在同一测序池中或在待测序的同一材料汇集中。
11.表观遗传靶区组可以通过各种方式分析。例如,在关于特定位置的修饰的可接受置信度低于关于序列可变靶区组中的准确性的可接受置信度的情况下(例如,在目的是了解在各基因座处的不同类型的修饰的频率而不必是修饰的确切位置的情况下),分析可以使用不依赖于靶内特定核苷酸的序列确定的高准确度的方法。实例包括确定修饰(诸如甲基化)的程度和/或片段的分布和尺寸,这可以指示从其获得片段的细胞中正常或异常的染色质结构。这样的分析可以通过测序进行,并且比确定存在或不存在序列突变(诸如碱基取代、插入或缺失)需要更少的数据(例如,序列读段的数目或测序覆盖率的深度)。
12.本公开内容旨在满足对无细胞dna的改进的分析的需要和/或提供其他益处。因此,提供以下示例性实施方案。
13.实施方案1是一种分析样品中dna的方法,所述方法包括:
14.a)将所述样品分区为多于一个子样品,所述多于一个子样品包括第一子样品和第二子样品,其中所述第一子样品包含比所述第二子样品具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的dna;
15.b)使所述第一子样品经历不同地影响所述第一子样品的dna中的第一核碱基和所述dna中的第二核碱基的程序,其中所述第一核碱基是修饰或未修饰的核碱基,所述第二核碱基是不同于所述第一核碱基的修饰或未修饰的核碱基,并且所述第一核碱基和所述第二
核碱基具有相同的碱基配对特异性;以及
16.c)以区分所述第一子样品的dna中的所述第一核碱基和所述第二核碱基的方式对所述第一子样品中的dna和所述第二子样品中的dna进行测序。
17.实施方案2是根据实施方案1所述的方法,其中所述dna包括从测试受试者获得的无细胞dna(cfdna)。
18.实施方案3是一种分析样品中的dna的方法,所述dna包括从测试受试者获得的无细胞dna(cfdna),所述方法包括:
19.a)将所述样品分区为多于一个子样品,所述多于一个子样品包括第一子样品和第二子样品,其中所述第一子样品包含比所述第二子样品具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的dna;
20.b)使所述第一子样品经历不同地影响所述第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序,其中所述第一核碱基是修饰或未修饰的核碱基,所述第二核碱基是不同于所述第一核碱基的修饰或未修饰的核碱基,并且所述第一核碱基和所述第二核碱基具有相同的碱基配对特异性;以及
21.c)以区分所述第一子样品的dna中的所述第一核碱基和所述第二核碱基的方式对至少所述第一子样品中的dna进行测序。
22.实施方案4是根据实施方案3所述的方法,其中步骤c)包括对至少所述第一子样品中的dna进行测序。
23.实施方案5是一种分析包含无细胞dna(cfdna)的样品的方法,所述方法包括:
24.a)将所述样品分区为多于一个子样品,所述多于一个子样品包括第一子样品和第二子样品,其中所述第一子样品包含比所述第二子样品具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的cfdna;
25.b)使所述第一子样品经历不同地影响所述第一子样品的cfdna中的第一核碱基和cfdna中的第二核碱基的程序,其中所述第一核碱基是修饰或未修饰的核碱基,所述第二核碱基是不同于所述第一核碱基的修饰或未修饰的核碱基,并且所述第一核碱基和所述第二核碱基具有相同的碱基配对特异性;
26.c)从所述第一子样品和所述第二子样品中捕获cfdna的至少表观遗传靶区组,
27.从而提供捕获的cfdna;以及
28.d)以区分来自所述第一子样品的cfdna中的所述第一核碱基和所述第二核碱基的方式对所述捕获的cfdna进行测序。
29.实施方案6是一种分析包含无细胞dna(cfdna)的样品的方法,所述方法包括:
30.a)将所述样品分区为多于一个子样品,所述多于一个子样品包括第一子样品和第二子样品,其中所述第一子样品包含比所述第二子样品具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的cfdna;
31.b)使所述第一子样品经历不同地影响所述第一子样品的cfdna中的第一核碱基和cfdna中的第二核碱基的程序,其中所述第一核碱基是修饰或未修饰的核碱基,所述第二核碱基是不同于所述第一核碱基的修饰或未修饰的核碱基,并且所述第一核碱基和所述第二核碱基具有相同的碱基配对特异性;
32.c)从所述第一子样品和所述第二子样品中捕获cfdna的多于一个靶区组,
33.其中所述多于一个靶区组包括序列可变靶区组和表观遗传靶区组,
34.从而提供捕获的cfdna;以及
35.d)以区分来自所述第一子样品的捕获的cfdna中的所述第一核碱基和所述第二核碱基的方式对所述捕获的cfdna进行测序。
36.实施方案7是根据实施方案6所述的方法,其中在所述样品中,对应于所述序列可变靶区组的cfdna分子以比对应于所述表观遗传靶区组的cfdna分子更大的捕获产量捕获。
37.实施方案8是一种从样品中分离无细胞dna(cfdna)的方法,所述方法包括:
38.a)将所述样品分区为多于一个子样品,所述多于一个子样品包括第一子样品和第二子样品,其中所述第一子样品包含比所述第二子样品具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的cfdna;
39.b)使所述第一子样品经历不同地影响所述第一子样品的cfdna中的第一核碱基和cfdna中的第二核碱基的程序,其中所述第一核碱基是修饰或未修饰的核碱基,所述第二核碱基是不同于所述第一核碱基的修饰或未修饰的核碱基,并且所述第一核碱基和所述第二核碱基具有相同的碱基配对特异性;
40.c)使所述第一子样品和所述第二子样品的cfdna与靶特异性探针组接触,
41.其中所述靶特异性探针组包含对序列可变靶组特异性的靶结合探针和对表观遗传靶组特异性的靶结合探针,
42.由此形成靶特异性探针与cfdna的复合物;
43.将所述复合物与未结合靶特异性探针的cfdna分离,从而提供对应于所述序列可变靶组的捕获的cfdna和对应于所述表观遗传靶组的捕获的cfdna;以及
44.d)以区分来自所述第一子样品的cfdna中的所述第一核碱基和所述第二核碱基的方式对所述捕获的cfdna进行测序。
45.实施方案9是根据实施方案8所述的方法,其中所述靶特异性探针组被配置为以比对应于所述表观遗传靶组的cfdna更大的捕获产量捕获对应于所述序列可变靶组的cfdna。
46.实施方案10是一种鉴定由肿瘤产生的dna的存在的方法,所述方法包括:
47.a)从测试受试者收集cfdna样品,
48.b)将所述cfdna样品分区为多于一个子样品,所述多于一个子样品包括第一子样品和第二子样品,其中所述第一子样品包含比所述第二子样品具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的捕获的cfdna;
49.c)使所述第一子样品经历不同地影响所述第一子样品的cfdna中的第一核碱基和cfdna中的第二核碱基的程序,其中所述第一核碱基是修饰或未修饰的核碱基,所述第二核碱基是不同于所述第一核碱基的修饰或未修饰的核碱基,并且所述第一核碱基和所述第二核碱基具有相同的碱基配对特异性;
50.d)从所述第一子样品和所述第二子样品中的cfdna捕获多于一个靶区组,
51.其中所述多于一个靶区组包括序列可变靶区组和表观遗传靶区组,从而提供包含捕获的cfdna的样品;以及
52.e)以区分所述第一子样品的cfdna中的所述第一核碱基和所述第二核碱基的方式对所述第一子样品中捕获的cfdna和所述第二子样品中捕获的cfdna进行测序。
53.实施方案11是根据实施方案10所述的方法,其中在所述样品中,对应于所述序列
可变靶区组的cfdna分子以比对应于所述表观遗传靶区组的cfdna分子更大的捕获产量捕获。
54.实施方案12是根据实施方案6-11中任一项所述的方法,所述方法包括将对应于所述序列可变靶区组的cfdna分子测序到比对应于所述表观遗传靶区组的cfdna分子更大的测序深度。
55.实施方案13是根据实施方案12所述的方法,其中将所述序列可变靶组的捕获的cfdna分子测序到所述表观遗传靶区组的捕获的cfdna分子的至少2倍大测序深度。
56.实施方案14是根据实施方案12所述的方法,其中将所述序列可变靶组的捕获的cfdna分子测序到所述表观遗传靶区组的捕获的cfdna分子的至少3倍大的测序深度。
57.实施方案15是根据实施方案12所述的方法,其中将所述序列可变靶组的捕获的cfdna分子测序到所述表观遗传靶区组的捕获的cfdna分子的4-10倍大的测序深度。
58.实施方案16是根据实施方案12所述的方法,其中将所述序列可变靶组的捕获的cfdna分子测序到所述表观遗传靶区组的捕获的cfdna分子的4-100倍大的测序深度。
59.实施方案17是根据实施方案6-16中任一项所述的方法,其中所述序列可变靶组的捕获的cfdna分子和所述表观遗传靶区组的捕获的cfdna分子在同一测序池中测序。
60.实施方案18是根据前述实施方案中任一项所述的方法,其中在测序之前扩增所述dna,或者其中所述方法包括捕获步骤并且在所述捕获步骤之前扩增所述dna。
61.实施方案19是根据实施方案5-18所述的方法,所述方法还包括在捕获之前将含条形码的衔接子连接到所述dna,任选地其中所述连接发生在扩增之前或与扩增同时发生。
62.实施方案20是根据实施方案5-19中任一项所述的方法,其中所述表观遗传靶区组包括高甲基化可变靶区组。
63.实施方案21是根据实施方案5-20中任一项所述的方法,其中所述表观遗传靶区组包括低甲基化可变靶区组。
64.实施方案22是根据实施方案20或21所述的方法,其中所述表观遗传靶区组包括甲基化对照靶区组。
65.实施方案23是根据实施方案5-22中任一项所述的方法,其中所述表观遗传靶区组包括片段化可变靶区组。
66.实施方案24是根据实施方案23所述的方法,其中所述片段化可变靶区组包括转录起始位点区。
67.实施方案25是根据实施方案23或24所述的方法,其中所述片段化可变靶区组包括ctcf结合区。
68.实施方案26是根据实施方案5-25中任一项所述的方法,其中捕获所述cfdna的多于一个靶区组包括使所述cfdna与对所述序列可变靶区组特异性的靶结合探针和对所述表观遗传靶区组特异性的靶结合探针接触。
69.实施方案27是根据实施方案26所述的方法,其中对所述序列可变靶区组特异性的靶结合探针以比对所述表观遗传靶区组特异性的靶结合探针更高的浓度存在。
70.实施方案28是根据实施方案26所述的方法,其中对所述序列可变靶区组特异性的靶结合探针以对所述表观遗传靶区组特异性的靶结合探针的至少2倍高的浓度存在。
71.实施方案29是根据实施方案26所述的方法,其中对所述序列可变靶区组特异性的
靶结合探针以对所述表观遗传靶区组特异性的靶结合探针的至少4倍或5倍高的浓度存在。
72.实施方案30是根据实施方案26所述的方法,其中对所述序列可变靶区组特异性的靶结合探针以对所述表观遗传靶区组特异性的靶结合探针的至少10倍、20倍、30倍、40倍、50倍、60倍、70倍、80倍、90倍或100倍高的浓度存在,或者其中对所述序列可变靶区组特异性的靶结合探针以对所述表观遗传靶区组特异性的靶结合探针浓度的2-3倍、3-4倍、4-5倍、5-7倍、7-10倍、10-20倍、20-30倍、30-40倍、40-50倍、50-60倍、60-70倍、70-80倍、80-90倍或90-100倍的范围内的浓度存在。
73.实施方案31是根据实施方案26-30中任一项所述的方法,其中对所述序列可变靶区组特异性的靶结合探针比对所述表观遗传靶区组特异性的靶结合探针具有更高的靶结合亲和力。
74.实施方案32是根据实施方案6-32中任一项所述的方法,其中所述表观遗传靶区组的足迹为所述序列可变靶区组的尺寸的至少2倍大。
75.实施方案33是根据实施方案32所述的方法,其中所述表观遗传靶区组的足迹为所述序列可变靶区组的尺寸的至少10倍大。
76.实施方案34是根据实施方案6-33中任一项所述的方法,其中所述序列可变靶区组的足迹为至少25kb或50kb。
77.实施方案35是根据前述实施方案中任一项所述的方法,其中将所述样品分区为多于一个子样品包括基于甲基化水平进行分区。
78.实施方案36是根据实施方案35所述的方法,其中所述分区步骤包括使收集的cfdna与固定在固体支持物上的甲基结合试剂接触。
79.实施方案37是根据前述实施方案中任一项所述的方法,所述方法包括对所述第一子样品和所述第二子样品差异性加标签。
80.实施方案38是根据实施方案37所述的方法,其中在使所述第一子样品经历不同地影响所述第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序之前,对所述第一子样品和所述第二子样品差异性加标签。
81.实施方案39是根据实施方案37或38所述的方法,其中在使所述第一子样品经历不同地影响所述第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序之后,汇集所述第一子样品和所述第二子样品。
82.实施方案40是根据实施方案37-39中任一项所述的方法,其中所述第一子样品和所述第二子样品在同一测序池中测序。
83.实施方案41是根据前述实施方案中任一项所述的方法,其中所述多于一个子样品包括第三子样品,所述第三子样品包含比所述第二子样品具有更大比例但比所述第一子样品具有更小比例的核苷酸修饰(例如,胞嘧啶修饰)的dna。
84.实施方案42是根据实施方案41所述的方法,其中所述方法还包括对所述第三子样品差异性加标签,以便使所述第三子样品与所述第一子样品和所述第二子样品可区分。
85.实施方案43是根据实施方案42所述的方法,其中在使所述第一子样品经历不同地影响所述第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序之后,将所述第一子样品、所述第二子样品和所述第三子样品组合,任选地其中所述第一子样品、所述第二子样品和所述第三子样品在同一测序池中测序。
86.实施方案44是根据前述实施方案中任一项所述的方法,其中所述第一子样品经历的所述程序改变所述第一核碱基的碱基配对特异性而基本不改变所述第二核碱基的碱基配对特异性。
87.实施方案45是根据前述实施方案中任一项所述的方法,其中所述第一核碱基是修饰或未修饰的胞嘧啶,并且所述第二核碱基是修饰或未修饰的胞嘧啶。
88.实施方案46是根据前述实施方案中任一项所述的方法,其中所述第一核碱基包括未修饰的胞嘧啶(c)。
89.实施方案47是根据前述实施方案中任一项所述的方法,其中所述第二核碱基包括5-甲基胞嘧啶(mc)。
90.实施方案48是根据前述实施方案中任一项所述的方法,其中所述第一子样品经历的所述程序包括亚硫酸氢盐转化。
91.实施方案49是根据实施方案1-46中任一项所述的方法,其中所述第一核碱基包括mc。
92.实施方案50是根据前述实施方案中任一项所述的方法,其中所述第二核碱基包括5-羟甲基胞嘧啶(hmc)。
93.实施方案51是根据实施方案50所述的方法,其中所述第一子样品经历的所述程序包括对5hmc的保护。
94.实施方案52是根据实施方案50所述的方法,其中所述第一子样品经历的所述程序包括tet辅助的亚硫酸氢盐转化。
95.实施方案53是根据实施方案50所述的方法,其中所述第一子样品经历的所述程序包括tet辅助的取代硼烷还原剂转化,任选地其中所述取代硼烷还原剂是2-甲基吡啶硼烷、吡啶硼烷、叔丁基胺硼烷或氨硼烷。
96.实施方案54是根据实施方案53所述的方法,其中所述取代硼烷还原剂是2-甲基吡啶硼烷或吡啶硼烷。
97.实施方案55是根据实施方案49-51或53-54中任一项所述的方法,其中所述第二核碱基包括c。
98.实施方案56是根据实施方案49-51或55中任一项所述的方法,其中所述第一子样品经历的所述程序包括对hmc的保护,随后用所述tet辅助的取代硼烷还原剂转化,任选地其中所述取代硼烷还原剂是2-甲基吡啶硼烷、吡啶硼烷、叔丁基胺硼烷或氨硼烷。
99.实施方案57是根据实施方案56所述的方法,其中所述取代硼烷还原剂是2-甲基吡啶硼烷或吡啶硼烷。
100.实施方案58是根据实施方案46、47、49-51或55中任一项所述的方法,其中所述第一子样品经历的所述程序包括对hmc的保护,随后经历mc和/或c的脱氨基。
101.实施方案59是根据实施方案58所述的方法,其中所述mc和/或c的脱氨基包括用aid/apobec家族dna脱氨基酶处理。
102.实施方案60是根据实施方案51或55-59中任一项所述的方法,其中对hmc的保护包括hmc的葡糖基化。
103.实施方案61是根据实施方案1-45、47、49或55中任一项所述的方法,其中所述第一子样品经历的所述程序包括化学辅助的取代硼烷还原剂转化,任选地其中所述取代硼烷还
原剂是2-甲基吡啶硼烷、吡啶硼烷、叔丁基胺硼烷或氨硼烷。
104.实施方案62是根据实施方案61所述的方法,其中所述取代硼烷还原剂是2-甲基吡啶硼烷或吡啶硼烷。
105.实施方案63是根据实施方案1-45、47、49、55或61-62中任一项所述的方法,其中所述第一核碱基包括hmc。
106.实施方案64是根据实施方案1-44中任一项所述的方法,其中所述第一子样品经历的所述程序包括将最初包含所述第一核碱基的dna与最初不包含所述第一核碱基的dna分离。
107.实施方案65是根据实施方案64所述的方法,其中所述第一核碱基是hmc。
108.实施方案66是根据实施方案64或65所述的方法,其中将最初包含所述第一核碱基的dna与最初不包含所述第一核碱基的dna分离包括标记所述第一核碱基。
109.实施方案67是根据实施方案66所述的方法,其中所述标记包括生物素化。
110.实施方案68是根据实施方案67所述的方法,其中所述标记包括葡糖基化。
111.实施方案69是根据实施方案68所述的方法,其中所述葡糖基化附接葡糖基叠氮基部分。
112.实施方案70是根据实施方案66或68所述的方法,其中所述标记包括葡糖基化,随后在生物素化之前将生物素部分附接到所述葡糖基。
113.实施方案71是根据实施方案70所述的方法,其中将生物素部分附接到所述葡糖基包括huisgen环加成化学。
114.实施方案72是根据实施方案54-61中任一项所述的方法,其中将最初包含所述第一核碱基的dna与最初不包含所述第一核碱基的dna分离包括使最初包含所述第一核碱基的dna与捕获剂结合。
115.实施方案73是根据实施方案72所述的方法,其中所述捕获剂包括生物素结合剂,任选地其中所述生物素结合剂包括亲和素或链霉亲和素。
116.实施方案74是根据实施方案64-73中任一项所述的方法,所述方法包括对最初包含所述第一核碱基的dna、最初不包含所述第一核碱基的dna和所述第二子样品的dna中的每一个差异性加标签。
117.实施方案75是根据实施方案74所述的方法,所述方法包括在差异性加标签之后汇集最初包含所述第一核碱基的dna、最初不包含所述第一核碱基的dna和所述第二子样品的dna,任选地其中最初包含所述第一核碱基的dna、最初不包含所述第一核碱基的dna和所述第二子样品的dna在同一测序池中测序。
118.实施方案76是根据实施方案1-44中任一项所述的方法,其中所述第一核碱基是修饰或未修饰的腺嘌呤,并且所述第二核碱基是修饰或未修饰的腺嘌呤。
119.实施方案77是根据实施方案1-44中任一项所述的方法,其中所述第一核碱基是修饰或未修饰的鸟嘌呤,并且所述第二核碱基是修饰或未修饰的鸟嘌呤。
120.实施方案78是根据实施方案1-44中任一项所述的方法,其中所述第一核碱基是修饰或未修饰的胸腺嘧啶,并且所述第二核碱基是修饰或未修饰的胸腺嘧啶。
121.实施方案79是根据前述实施方案中任一项所述的方法,其中所述第二亚群不经历不同地影响所述第一核碱基和所述第二核碱基的程序。
122.实施方案80是一种组合,所述组合包含捕获的dna的第一群体和第二群体,其中所述第一群体包含或源自比第二群体具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的dna,并且其中在碱基配对特异性改变之前最初存在于所述dna中的所述第一核碱基的形式是修饰或未修饰的核碱基,所述第二核碱基是不同于所述第一核碱基的修饰或未修饰的核碱基,并且在碱基配对特异性改变之前最初存在于所述dna中的所述第一核碱基的形式和所述第二核碱基具有相同的碱基配对特异性,并且所述第二群体不包含具有改变的碱基配对特异性的最初存在于所述dna中的所述第一核碱基的形式。
123.实施方案81是根据实施方案80所述的组合,其中所述第一群体包含选自第一组一个或更多个序列标签的序列标签并且所述第二群体包含选自第二组一个或更多个序列标签的序列标签,并且所述第二组序列标签不同于所述第一组序列标签。
124.实施方案82是根据实施方案81所述的组合,其中所述序列标签包括条形码。
125.实施方案83是根据实施方案80-82中任一项所述的组合,其中所述胞嘧啶修饰是甲基化。
126.实施方案84是根据实施方案80-83中任一项所述的组合,其中所述第一核碱基是修饰或未修饰的胞嘧啶,并且所述第二核碱基是修饰或未修饰的胞嘧啶。
127.实施方案85是根据实施方案80-84中任一项所述的组合,其中所述第一核碱基包括未修饰的胞嘧啶(c)。
128.实施方案86是根据实施方案80-85中任一项所述的组合,其中所述第二核碱基包括5-甲基胞嘧啶(mc)和5-羟甲基胞嘧啶(hmc)中的一种或两者。
129.实施方案87是根据实施方案80-86中任一项所述的组合,其中所述第一群体经历亚硫酸氢盐转化。
130.实施方案88是根据实施方案80-86中任一项所述的组合,其中所述第一核碱基包括mc。
131.实施方案89是根据实施方案80-88中任一项所述的组合,其中所述第二核碱基包括hmc。
132.实施方案90是根据实施方案80-89中任一项所述的组合,其中所述第一群体包含受保护的hmc。
133.实施方案91是根据实施方案84或90所述的组合,其中所述第一群体经历tet辅助的亚硫酸氢盐转化。
134.实施方案92是根据实施方案84或90所述的组合,其中所述第一群体经历tet辅助的取代硼烷还原剂转化,任选地其中所述取代硼烷还原剂是2-甲基吡啶硼烷、吡啶硼烷、叔丁基胺硼烷或氨硼烷。
135.实施方案93是根据实施方案90所述的组合,其中所述第一群体经历对hmc的保护,随后经历tet辅助的取代硼烷还原剂转化,任选地其中所述取代硼烷还原剂是2-甲基吡啶硼烷、吡啶硼烷、叔丁基胺硼烷或氨硼烷。
136.实施方案94是根据实施方案80-82、86、88-90或92-93中任一项所述的组合,其中所述第二核碱基包括c。
137.实施方案95是根据实施方案90所述的组合,其中所述第一群体经历对hmc的保护,随后经历mc和/或c的脱氨基。
138.实施方案96是根据实施方案90-95中任一项所述的组合,其中受保护的hmc包括葡糖基化的hmc。
139.实施方案97是根据实施方案80-83中任一项所述的组合,其中所述第一核碱基包括hmc。
140.实施方案98是根据实施方案80-83或97中任一项所述的组合,其中所述第二核碱基包括mc。
141.实施方案99是根据实施方案80-83或97-98中任一项所述的组合,其中所述第二核碱基包括c。
142.实施方案100是根据实施方案80-83或97-99中任一项所述的组合,其中所述第一群体经历化学辅助的取代硼烷还原剂转化,任选地其中所述取代硼烷还原剂是2-甲基吡啶硼烷、吡啶硼烷、叔丁基胺硼烷或氨硼烷。
143.实施方案101是根据实施方案80-83中任一项所述的组合,其中所述第一核碱基是修饰或未修饰的腺嘌呤,并且所述第二核碱基是修饰或未修饰的腺嘌呤。
144.实施方案102是根据实施方案80-83中任一项所述的组合,其中所述第一核碱基是修饰或未修饰的鸟嘌呤,并且所述第二核碱基是修饰或未修饰的鸟嘌呤。
145.实施方案103是根据实施方案80-83中任一项所述的组合,其中所述第一核碱基是修饰或未修饰的胸腺嘧啶,并且所述第二核碱基是修饰或未修饰的胸腺嘧啶。
146.实施方案104是一种组合,所述组合包含捕获的dna的第一群体和第二群体,其中:
147.所述第一群体包含或源自比所述第二群体具有更大比例的核苷酸修饰(例如,胞嘧啶修饰)的dna;
148.所述第一群体包括第一亚群和第二亚群;
149.所述第一亚群包含比所述第二亚群更大比例的第一核碱基;
150.其中所述第一核碱基是修饰或未修饰的核碱基,第二核碱基是不同于所述第一核碱基的修饰或未修饰的核碱基,并且所述第一核碱基和所述第二核碱基具有相同的碱基配对特异性;并且如果第一核碱基是修饰或未修饰的胸腺嘧啶,则第二核碱基是修饰或未修饰的胸腺嘧啶,并且所述第二群体不包含所述第一核碱基。
151.实施方案105是根据实施方案104所述的组合,其中所述第一核碱基是修饰或未修饰的胞嘧啶,并且所述第二核碱基是修饰或未修饰的胞嘧啶。
152.实施方案106是根据实施方案105所述的组合,其中所述第一核碱基是受保护的修饰的胞嘧啶。
153.实施方案107是根据实施方案105或106所述的组合,其中所述第一核碱基是hmc的衍生物。
154.实施方案108是根据实施方案107所述的组合,其中所述第一核碱基是葡糖基化的hmc。
155.实施方案109是根据实施方案107或108所述的组合,其中所述第一核碱基是生物素化的hmc。
156.实施方案110是根据实施方案106-109中任一项所述的组合,其中所述第一核碱基是β-6-叠氮基-葡糖基-5-羟甲基胞嘧啶的huisgen环加成产物,所述产物包含亲和标记。
157.实施方案111是根据实施方案104所述的组合,其中所述第一核碱基是修饰或未修
饰的腺嘌呤,并且所述第二核碱基是修饰或未修饰的腺嘌呤。
158.实施方案112是根据实施方案104所述的组合,其中所述第一核碱基是修饰或未修饰的鸟嘌呤,并且所述第二核碱基是修饰或未修饰的鸟嘌呤。
159.实施方案113是根据实施方案104所述的组合,其中所述第一核碱基是修饰或未修饰的胸腺嘧啶,并且所述第二核碱基是修饰或未修饰的胸腺嘧啶。
160.实施方案114是根据实施方案104-113中任一项所述的组合,其中所述第一亚群包含第一序列标签,所述第二亚群包含不同于所述第一序列标签的第二序列标签,并且所述第二群体包含不同于所述第一序列标签和所述第二序列标签的第三序列标签,任选地其中所述第一标签、所述第二标签和/或所述第三标签是条形码。
161.实施方案115是根据实施方案80-114中任一项所述的组合,其中所述捕获的dna包括cfdna。
162.实施方案116是根据实施方案80-115中任一项所述的组合,其中所述捕获的dna包含序列可变靶区和表观遗传靶区,并且所述序列可变靶区的浓度大于所述表观遗传靶区的浓度,其中所述浓度针对所述序列可变靶区和所述表观遗传靶区的足迹尺寸进行归一化。
163.实施方案117是根据实施方案116所述的组合,其中所述序列可变靶区的浓度为所述表观遗传靶区的浓度的至少2倍大。
164.实施方案118是根据实施方案116所述的组合,其中所述序列可变靶区的浓度为所述表观遗传靶区的浓度的至少4倍或5倍大。
165.实施方案119是根据实施方案116-118中任一项所述的组合,其中所述浓度是针对所述靶区的足迹尺寸进行归一化的质量/体积浓度。
166.实施方案120是根据实施方案116-119中任一项所述的组合,其中所述表观遗传靶区包括高甲基化可变靶区、低甲基化可变靶区、转录起始位点区和ctcf结合区中的一种、两种、三种或四种;任选地其中所述表观遗传靶区还包括甲基化对照靶区。
167.实施方案121是根据实施方案80-120中任一项所述的组合,所述组合根据实施方案1-79中任一项所述的方法产生。
168.实施方案122是一种系统,所述系统包括:
169.通信接口,所述通信接口通过通信网络接收由核酸测序仪从对根据实施方案1-79中任一项所述的方法的第一子样品中的dna和第二子样品中的dna进行测序生成的多于一个序列读段;和
170.控制器,所述控制器包括计算机可读介质或能够访问计算机可读介质,所述计算机可读介质包含非暂时性计算机可执行指令,当所述非暂时性计算机可执行指令由至少一个电子处理器执行时,进行包括以下的方法:
171.(i)通过所述通信网络接收由所述核酸测序仪生成的所述序列读段;和
172.(ii)将所述多于一个序列读段映射到一个或更多个参考序列以生成映射的序列读段。
173.实施方案123是一种系统,所述系统包括:
174.通信接口,所述通信接口通过通信网络接收由核酸测序仪从对根据实施方案80-121中任一项所述的捕获的dna的第一群体和第二群体的组合进行测序生成的多于一个序列读段;和
175.控制器,所述控制器包括计算机可读介质或能够访问计算机可读介质,所述计算机可读介质包含非暂时性计算机可执行指令,当所述非暂时性计算机可执行指令由至少一个电子处理器执行时,进行包括以下的方法:
176.(i)通过所述通信网络接收由所述核酸测序仪生成的所述序列读段;和
177.(ii)将所述多于一个序列读段映射到一个或更多个参考序列以生成映射的序列读段。
178.实施方案124是根据实施方案122或123所述的系统,其中由所述至少一个电子处理器执行的所述方法还包括:
179.(iii)处理对应于所述序列可变靶区组和所述表观遗传靶区组的映射的序列读段,以确定所述受试者患有癌症的可能性。
180.实施方案125是根据实施方案1-79中任一项所述的方法,所述方法还包括确定所述受试者患有癌症的可能性。
181.实施方案126是根据前一项实施方案所述的方法,其中所述测序生成多于一个测序读段;并且所述方法还包括将所述多于一个序列读段映射到一个或更多个参考序列以生成映射的序列读段,以及处理对应于所述序列可变靶区组和所述表观遗传靶区组的所述映射的序列读段以确定所述受试者患有癌症的可能性。
182.实施方案127是根据实施方案1-79中任一项所述的方法,其中所述测试受试者先前被诊断为患有癌症并接受了一个或更多个先前的癌症治疗,任选地其中在所述一个或更多个先前的癌症治疗之后的一个或更多个预选时间点获得所述cfdna。
183.实施方案128是根据前一项实施方案所述的方法,所述方法还包括对cfdna分子的捕获组进行测序,由此产生序列信息组。
184.实施方案129是根据前一项实施方案所述的方法,其中将所述序列可变靶区组的捕获的dna分子测序到比所述表观遗传靶区组的捕获的dna分子更大的测序深度。
185.实施方案130是根据实施方案128或129所述的方法,所述方法还包括使用所述序列信息组在预选时间点检测来源于或源自肿瘤细胞的dna的存在或不存在。
186.实施方案131是根据前一项实施方案所述的方法,所述方法还包括确定癌症复发评分,所述癌症复发评分指示来源于或源自所述测试受试者的肿瘤细胞的dna的存在或不存在。
187.实施方案132是根据前一项实施方案所述的方法,所述方法还包括基于所述癌症复发评分确定癌症复发状态,其中当癌症复发评分被确定为处于或高于预定阈值时,将所述测试受试者的癌症复发状态确定为处于癌症复发风险,或者当所述癌症复发评分低于所述预定阈值时,将所述测试受试者的癌症复发状态确定为处于较低的癌症复发风险。
188.实施方案133是根据实施方案131或132所述的方法,所述方法还包括将所述测试受试者的癌症复发评分与预定癌症复发阈值进行比较,并且当所述癌症复发评分高于所述癌症复发阈值时,将所述测试受试者分类为后续癌症治疗的候选者,或者当所述癌症复发评分低于所述癌症复发阈值时,将所述测试受试者分类为后续癌症治疗的非候选者。
189.实施方案134是根据实施方案131-133中任一项所述的方法,其中所述测试受试者处于癌症复发的风险,并且被分类为后续癌症治疗的候选者。
190.实施方案135是根据实施方案131、133或134中任一项所述的方法,其中所述后续
癌症治疗包括化学疗法或施用治疗组合物。
191.实施方案136是根据实施方案132-135中任一项所述的方法,其中所述来源于或源自肿瘤细胞的dna是无细胞dna。
192.实施方案137是根据实施方案132-135中任一项所述的方法,其中所述来源于或源自肿瘤细胞的dna是从组织样品中获得的。
193.实施方案138是根据实施方案129-137中任一项所述的方法,所述方法还包括基于所述癌症复发评分确定所述测试受试者的无疾病存活(dfs)期。
194.实施方案139是根据实施方案138所述的方法,其中dfs期为1年、2年、3年、4年、5年或10年。
195.实施方案140是根据实施方案128-139中任一项所述的方法,其中所述序列信息组包括序列可变靶区序列,并且确定癌症复发评分包括确定至少第一子评分,所述第一子评分指示序列可变靶区序列中存在的snv、插入/缺失、cnv和/或融合的量。
196.实施方案141是根据实施方案140所述的方法,其中所述序列可变靶区中选自1个、2个、3个、4个或5个的突变数目足以使所述第一子评分导致癌症复发评分被分类为癌症复发阳性,任选地其中所述突变数目选自1个、2个或3个。
197.实施方案142是根据实施方案128-141中任一项所述的方法,其中所述序列信息组包括表观遗传靶区序列,并且确定癌症复发评分包括确定第二子评分,所述第二子评分指示所述表观遗传靶区序列中异常序列读段的量。
198.实施方案143是根据实施方案142所述的方法,其中异常序列读段包括指示高甲基化可变靶序列的甲基化的序列读段和/或指示片段化可变靶区中异常片段化的序列读段。
199.实施方案144是根据实施方案143所述的方法,其中大于或等于0.001%-10%的范围内的值的指示高甲基化可变靶区组中的高甲基化和/或片段化可变靶区组中的异常片段化的对应于高甲基化可变靶区组和/或片段化可变靶区组的读段比例足以使所述第二子评分被分类为癌症复发阳性。
200.实施方案145是根据实施方案144所述的方法,其中所述范围为0.001%-1%或0.005%-1%。
201.实施方案146是根据实施方案144所述的方法,其中所述范围为0.01%-5%或0.01%-2%。
202.实施方案147是根据实施方案144所述的方法,其中所述范围为0.01%-1%。
203.实施方案148是根据实施方案128-147中任一项所述的方法,所述方法还包括从指示一个或更多个指示来源于肿瘤细胞的特征的序列信息组中的读段分数确定肿瘤dna分数。
204.实施方案149是根据实施方案148所述的方法,其中所述一个或更多个指示来源于肿瘤细胞的特征包括以下中的一个或更多个:序列可变靶区中的改变、高甲基化可变靶区的高甲基化和片段化可变靶区的异常片段化。
205.实施方案150是根据实施方案148或149所述的方法,所述方法还包括至少部分地基于所述肿瘤dna分数来确定癌症复发评分,其中大于或等于10-11
至1或10-10
至1的范围内的预定值的肿瘤dna分数足以使所述癌症复发评分被分类为癌症复发阳性。
206.实施方案151是根据实施方案150所述的方法,其中大于或等于10-10
至10-9
、10-9
至
10-8
、10-8
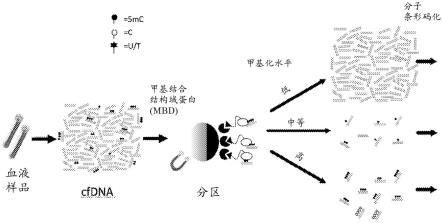
至10-7
、10-7
至10-6
、10-6
至10-5
、10-5
至10-4
、10-4
至10-3
、10-3
至10-2
或10-2
至10-1
的范围内的预定值的肿瘤dna分数足以使所述癌症复发评分被分类为癌症复发阳性。
207.实施方案152是根据实施方案150或151所述的方法,其中所述预定值在10-8
至10-6
的范围内或者是10-7
。
208.实施方案153是根据实施方案149-152中任一项所述的方法,其中如果所述肿瘤dna分数大于或等于所述预定值的累积概率为至少0.5、0.75、0.9、0.95、0.98、0.99、0.995或0.999,则将所述肿瘤dna分数确定为大于或等于所述预定值。
209.实施方案154是根据实施方案153所述的方法,其中所述累积概率为至少0.95。
210.实施方案155是根据实施方案153所述的方法,其中所述累积概率在0.98-0.995的范围内或者是0.99。
211.实施方案156是根据实施方案128-155中任一项所述的方法,其中所述序列信息组包括序列可变靶区序列和表观遗传靶区序列,并且确定癌症复发评分包括确定指示序列可变靶区序列中存在的snv、插入/缺失、cnv和/或融合的量的第一子评分和指示表观遗传靶区序列中异常序列读段的量的第二子评分,以及组合所述第一子评分和所述第二子评分以提供所述癌症复发评分。
212.实施方案157是根据实施方案156所述的方法,其中组合所述第一子评分和所述第二子评分包括将阈值独立地应用于每个子评分(例如,在序列可变靶区中大于预定的突变数目(例如,》1),并且在表观遗传靶区中大于预定的异常(例如,肿瘤)读段分数),或者训练机器学习分类器以基于多于一个阳性训练样品和阴性训练样品来确定状态。
213.实施方案158是根据实施方案157所述的方法,其中在-4至2或-3至1的范围内的组合评分的值足以使所述癌症复发评分被分类为癌症复发阳性。
214.实施方案159是根据实施方案127-158中任一项所述的方法,其中所述一个或更多个预选时间点选自由以下组成的组:在施用所述一个或更多个先前的癌症治疗之后的1个月、2个月、3个月、4个月、5个月、6个月、7个月、8个月、9个月、10个月、11个月、1年、1.5年、2年、3年、4年和5年。
215.实施方案160是根据实施方案127-159中任一项所述的方法,其中所述癌症是结肠直肠癌。
216.实施方案161是根据实施方案127-160中任一项所述的方法,其中所述一个或更多个先前的癌症治疗包括手术。
217.实施方案162是根据实施方案127-161中任一项所述的方法,其中所述一个或更多个先前的癌症治疗包括施用治疗组合物。
218.实施方案163是根据实施方案127-162中任一项所述的方法,其中所述一个或更多个先前的癌症治疗包括化学疗法。
219.实施方案164是根据实施方案80-120中任一项所述的组合,其中所述改变的碱基配对特异性是通过化学转化产生的。
220.实施方案165是根据实施方案164所述的组合,其中所述化学转化选自由以下组成的组:(i)亚硫酸氢盐转化,(ii)tet辅助的亚硫酸氢盐转化,(iii)tet辅助的取代硼烷还原剂转化,和(iv)对hmc的保护,随后为tet辅助的取代硼烷还原剂转化。
221.i.附图简述
222.图1说明根据本公开内容从血液样品开始的某些实施方案的示例性工作流程,其中从血液样品中分离cfdna;使用甲基结合结构域蛋白(mbd)将cfdna分区为低、中等和高甲基化子样品;使每个子样品经历分子条形码化以对来自低、中等和高甲基化子样品的dna可区分地加标签;将高甲基化子样品用亚硫酸氢盐(bs)处理,将c转化为u;并且将样品(以任何合适的顺序)汇集、捕获、扩增和测序。通常类似于本示例性方法的方法包括第一子样品(其为高甲基化子样品)的亚硫酸氢盐转化,通过转化或不存在转化指示哪些胞嘧啶位置是或不是未修饰的胞嘧啶(对比mc或hmc)。
223.图2说明根据本公开内容从血液样品开始的某些实施方案的示例性工作流程,其中从血液样品中分离cfdna;使用甲基结合结构域蛋白(mbd)将cfdna分区为低、中等和高甲基化子样品;使每个子样品经历分子条形码化以对来自低、中等和高甲基化子样品的dna可区分地加标签;将高甲基化子样品用β-葡糖基转移酶(经由葡糖基化保护hmc)和apobec3a(a3a)处理(使c和mc脱氨基使得它们将被测序为u/t);并且将样品(以任何合适的顺序)汇集、捕获、扩增和测序。通常类似于本示例性方法的方法包括第一子样品(其为高甲基化子样品)的hmc保护和mc脱氨基,通过转化或不存在转化指示哪些胞嘧啶位置是或不是未修饰的胞嘧啶或mc(对比hmc)。
224.图3说明根据本公开内容从血液样品开始的某些实施方案的示例性工作流程,其中从血液样品中分离cfdna;使用甲基结合结构域蛋白(mbd)将cfdna分区为低、中等和高甲基化子样品;使用hmc-seal进一步分离高甲基化子样品,所述hmc-seal包括用βgt处理和生物素化,从而将含hmc的dna与其他dna分离;使每个子样品经历分子条形码化以对来自低甲基化、中等甲基化、高甲基化和高甲基化 hmc子样品的dna可区分地加标签;并且将样品(以任何合适的顺序)汇集、捕获、扩增和测序。通常类似于本示例性方法的方法包括将高甲基化子样品中含hmc的dna与其他dna分离,指示哪些高甲基化dna的读段含有hmc。
225.图4是适用于本公开内容的一些实施方案的系统的实例的示意图。
226.图5示出如实施例2中描述的从患有早期结肠直肠癌的受试者(“早期crc”)和三名健康受试者(“正常”)的三个样品获得的cpg甲基化定量结果。对于早期crc图,maf指示突变等位基因分数。
227.ii.某些实施方案的详细描述
228.现在将详细述及本发明的某些实施方案。虽然将结合这样的实施方案描述本发明,但是应当理解它们并非意图本发明限于这些实施方案。相反,意图本发明覆盖所有替代、修改和等同方案,它们可以被包括在由所附的权利要求书限定的本发明内。
229.在详细描述本教导之前,应理解本公开内容不限于特定组合物或过程步骤,因为其可以变化。应注意的是,如本说明书和所附的权利要求书中使用的,单数形式“一个/种(a)”、“一个/种(an)”和“该/所述(the)”包括复数指代物,除非上下文另外明确规定。因此,例如,提及“核酸”包括多于一个核酸,提及“细胞”包括多于一个细胞,等等。
230.数值范围包括限定该范围的数字。考虑到与测量相关的有效数字和误差,测量值和可测量值应被理解为是近似的。此外,“包含(comprise)”、“包含(comprises)”、“包含(comprising)”、“含有(contain)”、“含有(contains)”、“含有(containing)”、“包括(include)”、“包括(includes)”和“包括(including)”的使用并非意图限制。应该理解,前述概括描述和详细描述两者仅是示例性的和说明性的,而不是限制本教导。
231.除非在以上说明书中具体提到,否则说明书中叙述的“包含/包括”各种组分/组件的实施方案也被设想为“由所叙述的组分/组件组成”或“主要由所叙述的组分/组件组成”;说明书中叙述的“由各种组分/组件组成”的实施方案也被设想为“包含/包括”或“主要由所叙述的组分/组件组成”;并且说明书中叙述的“主要由各种组分/组件组成”的实施方案也被设想为“由所叙述的组分/组件组成”或“包含/包括”所叙述的组分/组件(这种可互换性不应用于权利要求书中对这些术语的使用)。
232.本文使用的章节标题用于组织目的,而不应被解释为以任何方式限制所公开的主题。在通过引用并入的任何文件或其他材料与本说明书的任何明确内容(包括定义)相矛盾的情况下,以本说明书为准。
233.a.定义
[0234]“无细胞dna”、“cfdna分子”或简单地“cfdna”包括在受试者中以细胞外形式(例如,在血液、血清、血浆或其他体液诸如淋巴、脑脊液、尿液或痰中)天然存在的dna分子。虽然cfdna最初存在于大型复杂生物有机体(例如,哺乳动物)中的一个或更多个细胞中,但cfdna经历了从细胞中释放到生物体中存在的流体中,并且可以通过获得流体的样品来获得,而不需要进行体外细胞裂解步骤。
[0235]
如本文使用的,当具有修饰或其他特征的核苷酸在第一子样品或群体中的分数高于在第二群体中时,该修饰或其他特征在核酸的第一子样品或群体中以比第二子样品或群体中“更大比例”呈现。例如,如果在第一子样品中,十分之一的核苷酸是mc,而在第二子样品中,二十分之一的核苷酸是mc,则第一子样品比第二子样品包含更大比例的5-甲基化的胞嘧啶修饰。
[0236]
如本文使用的,“基本不改变特定核碱基的碱基配对特异性”意指包含可测序的该核碱基的大多数分子不具有相对于该核碱基在其最初分离的样品中的碱基配对特异性的第二核碱基的碱基配对特异性的改变。在一些实施方案中,75%、90%、95%或99%的分子不具有相对于该核碱基在其最初分离的样品中的碱基配对特异性的第二核碱基的碱基配对特异性的改变。
[0237]
如本文使用的,“碱基配对特异性”是指特定碱基最优先配对的标准dna碱基(a、c、g或t)。因此,例如,未修饰的胞嘧啶和5-甲基胞嘧啶具有相同的碱基配对特异性(即,对g的特异性),而尿嘧啶和胞嘧啶具有不同的碱基配对特异性,因为尿嘧啶对a具有碱基配对特异性,而胞嘧啶对g具有碱基配对特异性。尿嘧啶与g形成摆动对(wobble pair)的能力是不重要的,因为无论如何,在四种标准dna碱基中,尿嘧啶最优先与a配对。
[0238]
如本文使用的,包含多于一个成员的“组合”是指包含成员的单个组合物或一组邻近的组合物,例如,在单独的容器或较大容器内的区室中,诸如在多孔板、管架、冰箱、冷冻器、培养箱、水浴、冰桶、机器或其他储存形式中。
[0239]
探针集合对特定靶组的“捕获产量”是指在典型条件下探针集合捕获的对应于该靶组的核酸的量(例如,相对于另一靶组的量或绝对量)。示例性的典型捕获条件是将样品核酸与探针在含有严格杂交缓冲液的小反应体积(约20μl)中在65℃孵育10-18小时。捕获产量可以用绝对值表示,或者对于多于一个探针集合用相对值表示。当对多于一个靶区组的捕获产量进行比较时,将它们针对靶区组的足迹尺寸(例如,以每千个碱基为基础)进行归一化。因此,例如,如果第一靶区和第二靶区的足迹尺寸分别为50kb和500kb(提供0.1的
归一化因子),则当对应于第一靶区组的捕获的dna的质量/体积浓度比对应于第二靶区组的捕获的dna的质量/体积浓度多0.1倍时,对应于第一靶区组的dna以比对应于第二靶区组的dna更高的产量被捕获。作为另一实例,使用同样的足迹尺寸,如果对应于第一靶区组的捕获的dna具有对应于第二靶区组的捕获的dna的质量/体积浓度的0.2倍的质量/体积浓度,则对应于第一靶区组的dna以对应于第二靶区组的dna两倍大的捕获产量被捕获。
[0240]“捕获”一个或更多个靶核酸是指优先将所述一个或更多个靶核酸与非靶核酸分离或分开。
[0241]
核酸的“捕获组(captured set)”是指已经历捕获的核酸。
[0242]“靶区组(target-region set)”或“靶区组(set of target regions)”是指被靶向捕获和/或被探针组靶向(例如,通过序列互补性)的多于一个基因组基因座。
[0243]“对应于靶区组”意指核酸诸如cfdna来源于靶区组中的基因座或特异性地结合针对该靶区组的一种或更多种探针。
[0244]
在探针或其他寡核苷酸与靶序列的上下文中的“特异性结合”意指在适当的杂交条件下,寡核苷酸或探针与它的靶序列或其复制物杂交,以形成稳定的探针:靶杂交体,而同时稳定的探针:非靶杂交体的形成最小化。因此,探针与靶序列或其复制物杂交的程度比与非靶序列杂交的程度大到足以实现对靶序列的捕获或检测。适当的杂交条件是本领域熟知的,可以基于序列组成来预测,或者可以通过使用常规测试方法来确定(参见,例如,sambrook等人,molecular cloning,a laboratory manual,第二版.(cold spring harbor laboratory press,cold spring harbor,ny,1989)在
§§
1.90-1.91,7.37-7.57,9.47-9.51和11.47-11.57,特别是
§§
9.50-9.51,11.12-11.13,11.45-11.47和11.55-11.57,通过引用并入本文)。
[0245]“序列可变靶区组”是指在赘生性细胞(例如,肿瘤细胞和癌细胞)中可能表现出序列改变(诸如核苷酸取代(即,单核苷酸变异)、插入、缺失或基因融合或转座)的靶区组。
[0246]“表观遗传靶区组”是指在赘生性细胞(例如,肿瘤细胞和癌细胞)中可能显示出不依赖于序列的改变或者在来自患有癌症的受试者的cfdna中可能显示出相对于来自健康受试者的cfdna的不依赖于序列的改变的靶区组。不依于赖序列的改变的实例包括但不限于甲基化(增加或降低)、核小体分布、ctcf结合、转录起始位点和调控蛋白结合区的改变。对于本发明的目的,易感于与赘生物、肿瘤或癌症相关的聚焦扩增(focal amplification)和/或基因融合的基因座也可以被包括在表观遗传靶区组中,因为通过测序检测拷贝数的改变或检测映射到参考基因组中多于一个基因座的融合序列趋向于比检测核苷酸取代、插入或缺失更类似于检测以上讨论的示例性表观遗传改变,例如,在以下方面:聚焦扩增和/或基因融合可以以相对浅的测序深度检测到,因为它们的检测不依赖于一个或几个单独位置处的碱基判定(call)的准确度。在一些实施方案中,表观遗传靶区组包括一个或更多个基因组区域,其中这些区中的cfdna分子的表观遗传状态(例如,甲基化状态)在癌症中不变,但是它们在血液中的存在/量指示来自某些组织(例如,癌症来源)的cfdna向循环中的增加的异常的呈现。
[0247]
如果核酸来源于肿瘤细胞,则该核酸是“由肿瘤产生的”或ctdna或循环肿瘤dna。肿瘤细胞是来源于肿瘤的赘生性细胞,不管它们是保留在肿瘤中还是变得与肿瘤分开(如在例如转移的癌细胞和循环的肿瘤细胞的情况下)。
[0248]
术语“甲基化”或“dna甲基化”是指向核酸分子中的核苷酸碱基添加甲基基团。在一些实施方案中,甲基化是指向cpg位点(胞嘧啶-磷酸-鸟嘌呤位点(即,在核酸序列的5
’→3’
方向胞嘧啶随后是鸟嘌呤)处的胞嘧啶添加甲基基团。在一些实施方案中,dna甲基化是指向腺嘌呤添加甲基基团,诸如n
6-甲基腺嘌呤中。在一些实施方案中,dna甲基化是5-甲基化(对胞嘧啶第5个碳的修饰)。在一些实施方案中,5-甲基化是指向胞嘧啶的5c位置添加甲基基团以生成5-甲基胞嘧啶(5mc)。在一些实施方案中,甲基化包括5mc的衍生物。5mc的衍生物包括但不限于5-羟甲基胞嘧啶(5-hmc)、5-甲酰基胞嘧啶(5-fc)和5-羧基胞嘧啶(5-cac)。在一些实施方案中,dna甲基化是3c甲基化(对胞嘧啶第3个碳的修饰)。在一些实施方案中,3c甲基化包括向胞嘧啶的3c位置添加甲基基团以生成3-甲基胞嘧啶(3mc)。甲基化也可以发生在非cpg位点处,例如,甲基化可以发生在cpa、cpt或cpc位点处。dna甲基化可以改变甲基化的dna区域的活性。例如,当启动子区中的dna被甲基化时,基因的转录可能被阻遏。dna甲基化对于正常发育是至关重要的,并且甲基化的异常可以破坏表观遗传调控。表观遗传调控的破坏,例如阻遏,可以引起疾病,诸如癌症。dna中的启动子甲基化可以指示癌症。
[0249]
术语“高甲基化(hypermethylation)”是指相对于核酸分子群体(例如,样品)内的其他核酸分子,核酸分子的增加的甲基化水平或程度。在一些实施方案中,高甲基化dna可以包括包含至少1个甲基化残基、至少2个甲基化残基、至少3个甲基化残基、至少5个甲基化残基或至少10个甲基化残基的dna分子。
[0250]
术语“低甲基化(hypomethylation)”是指相对于核酸分子群体(例如,样品)内的其他核酸分子,核酸分子的降低的甲基化水平或程度。在一些实施方案中,低甲基化dna包括未甲基化的dna分子。在一些实施方案中,低甲基化dna可以包括包含0个甲基化残基、至多1个甲基化残基、至多2个甲基化残基、至多3个甲基化残基、至多4个甲基化残基或至多5个甲基化残基的dna分子。
[0251]
如本文使用的术语“或其组合(or a combination thereof)”和“或其组合(or combinations thereof)”是指该术语之前所列术语的任何和所有排列和组合。例如,“a、b、c或其组合”旨在包括以下中的至少一种:a、b、c、ab、ac、bc或abc,并且如果顺序在特定上下文中是重要的,则还包括ba、ca、cb、acb、cba、bca、bac或cab。继续这个实例,明确包括了含有一个或更多个项目或术语的重复的组合,诸如bb、aaa、aab、bbc、aaabcccc、cbbaaa、cababb等。本领域技术人员将理解,通常不存在对任何组合中的项目或术语的数目限制,除非另外从上下文明显。
[0252]“或”以包含性意义使用,即等同于“和/或”,除非上下文另有要求。
[0253]
b.示例性方法
[0254]
1.将样品分区为多于一个子样品;样品的方面;表观遗传特征分析
[0255]
在本文描述的某些实施方案中,可以基于核酸的一个或更多个特征对不同形式的核酸群体(例如,样品中的高甲基化dna和低甲基化dna,诸如如本文描述的cfdna的捕获组)进行物理分区,然后进一步分析,例如,差异修饰或分离核碱基、加标签和/或测序。这种方法可以用来确定,例如,某些序列是高甲基化的还是低甲基化的。在一些实施方案中,对高甲基化可变表观遗传靶区进行分析以确定它们是否显示出肿瘤细胞的高甲基化特征,和/或对低甲基化可变表观遗传靶区进行分析以确定它们是否显示出肿瘤细胞的低甲基化特
征。另外,通过将异质性核酸群体分区,人们可以增加罕见信号,例如,通过富集在群体的一种级分(或一个分区)中更普遍的罕见核酸分子。例如,通过将样品分区为高甲基化和低甲基化核酸分子,可以更容易地检测出存在于高甲基化dna中但在低甲基化dna中较少(或不存在)的遗传变异。通过分析样品的多于一个级分,可以对基因组的单个基因座或核酸种类进行多维分析,并因此可以实现更大的灵敏度。
[0256]
在一些情况下,将异质性核酸样品分区为两个或更多个分区(例如,至少3个、4个、5个、6个或7个分区)。在一些实施方案中,对每个分区差异性加标签。然后,可以将加标签的分区汇集在一起,用于集体样品制备和/或测序。分区-加标签-汇集步骤可以发生多于一次,其中每一轮分区基于不同的特征(本文提供的实例)发生,并使用区别于其他分区和分区手段的差异性标签来加标签。
[0257]
可以用于分区的特征的实例包括序列长度、甲基化水平、核小体结合、序列错配、免疫沉淀和/或与dna结合的蛋白。所得的分区可以包括以下核酸形式中的一种或更多种:单链dna(ssdna)、双链dna(dsdna)、较短dna片段和较长dna片段。在一些实施方案中,通常进行基于胞嘧啶修饰(例如,胞嘧啶甲基化)或甲基化的分区,并且任选地与至少一个另外的分区步骤组合,该步骤可以基于dna的任何前述特征或形式。在一些实施方案中,将异质性核酸群体分区为具有一个或更多个表观遗传修饰和不具有所述一个或更多个表观遗传修饰的核酸。表观遗传修饰的实例包括甲基化的存在或不存在、甲基化水平、甲基化类型(例如,5-甲基胞嘧啶与其他类型的甲基化,诸如腺嘌呤甲基化和/或胞嘧啶羟甲基化)、以及与一种或更多种蛋白(诸如组蛋白)的缔合和缔合水平。可选地或另外地,可以将异质性核酸群体分区为与核小体缔合的核酸分子和不含核小体的核酸分子。可选地或另外地,可以将异质性核酸群体分区为单链dna(ssdna)和双链dna(dsdna)。可选地或另外地,异质性核酸群体可以基于核酸长度(例如,最大160bp的分子和具有大于160bp的长度的分子)来分区。
[0258]
在一些情况下,将每个分区(代表不同的核酸形式)差异性标记,并将分区汇集在一起,然后测序。在其他情况下,将不同的形式单独测序。
[0259]
在一些实施方案中,将不同核酸群体分区为两个或更多个不同的分区。每个分区代表不同的核酸形式,并且第一分区(也称为子样品)包含比第二子样品具有更大比例的胞嘧啶修饰的dna。将每个分区不同地加标签。使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序,其中第一核碱基是修饰或未修饰的核碱基,第二核碱基是不同于第一核碱基的修饰或未修饰的核碱基,并且第一核碱基和第二核碱基具有相同的碱基配对特异性。将加标签的核酸汇集在一起,然后测序。获得序列读段并进行分析,包括以计算机(in silico)区分第一子样品的dna中的第一核碱基和第二核碱基。将标签用于分选来自不同分区的读段。可以在各个分区的水平以及整个核酸群体水平上进行分析以检测遗传变异。例如,分析可以包括以计算机分析来确定遗传变异,诸如每个分区的核酸中的cnv、snv、插入/缺失、融合。在一些情况下,以计算机分析可以包括确定染色质结构。例如,序列读段的覆盖率可以用来确定核小体在染色质中的定位。较高的覆盖率可以与基因组区域中较高的核小体占据度相关联,而较低的覆盖率可以与较低的核小体占据度或核小体缺失区(nucleosome depleted region,ndr)相关联。
[0260]
样品可以包括不同修饰的核酸,包括对核苷酸的复制后修饰和与一个或更多个蛋
白的结合(通常是非共价的)。
[0261]
在实施方案中,核酸群体是从怀疑患有赘生物、肿瘤或癌症或先前被诊断为患有赘生物、肿瘤或癌症的受试者的血清、血浆或血液样品获得的核酸群体。核酸群体包括具有不同甲基化水平的核酸。甲基化可以由任何一个或更多个复制后或转录后修饰发生。复制后修饰包括对核苷酸胞嘧啶的修饰,特别是在核碱基的5-位置处,例如5-甲基胞嘧啶、5-羟甲基胞嘧啶、5-甲酰基胞嘧啶和5-羧基胞嘧啶。
[0262]
亲和剂可以是具有期望的特异性的抗体、天然结合配偶体或其变体(bock等人,nat biotech 28:1106-1114(2010);song等人,nat biotech 29:68-72(2011)),或例如通过噬菌体展示选择的对特定靶具有特异性的人工肽。
[0263]
本文设想的捕获部分的实例包括如本文描述的甲基结合结构域(mbd)和甲基结合蛋白(mbp),包括蛋白诸如mecp2和优先与5-甲基胞嘧啶结合的抗体。
[0264]
同样,对不同形式核酸的分区可以使用组蛋白结合蛋白进行,该组蛋白结合蛋白可以分离与组蛋白结合的核酸与游离或未结合的核酸。可以用于本文公开的方法的组蛋白结合蛋白的实例包括rbbp4、rbap48和sant结构域肽。
[0265]
对于一些亲和剂和修饰,尽管与剂的结合可以取决于核酸是否带有修饰而以基本上全或无的方式发生,但是分离可以是一定程度的。在这样的情况下,与修饰未被充分代表的核酸(nucleic acids underrepresented in the modification)相比,修饰被过度代表的核酸(nucleic acids overrepresented in a modification)与剂以更大的程度与剂结合。可选地,具有修饰的核酸可以以全或无的方式结合。但是然后,各种水平的修饰可以从结合剂顺序洗脱。
[0266]
例如,在一些实施方案中,分区可以是二元的或者基于修饰的程度/水平。例如,可以使用甲基结合结构域蛋白(例如methylminer甲基化dna富集试剂盒(thermofisher scientific))将所有甲基化片段与未甲基化的片段分区。随后,另外的分区可以包括通过调整含有甲基结合结构域和结合片段的溶液的盐浓度来洗脱具有不同甲基化水平的片段。随着盐浓度增加,具有更大甲基化水平的片段被洗脱。
[0267]
在一些情况下,最终分区代表具有不同程度的修饰(过度代表性(over representative)或代表性不足(under representative)的修饰)的核酸。过度代表性和代表性不足可以由核酸带有的修饰的数目相对于群体中每条链的修饰的中位数来定义。例如,如果样品中的核酸中5-甲基胞嘧啶残基的中位数为2,则包含多于两个5-甲基胞嘧啶残基的核酸的该修饰是过度代表性的,而具有1个或0个5-甲基胞嘧啶残基的核酸是代表性不足的。亲和分离的作用是富集结合相中修饰被过度代表的核酸和非结合相(即,溶液中)中修饰未被充分代表的核酸。结合相的核酸可以在后续处理之前洗脱。
[0268]
当使用methylminer甲基化dna富集试剂盒(thermofisher scientific)时,可以使用顺序洗脱将不同水平的甲基化分区。例如,可以通过使核酸群体与来自试剂盒的附接至磁珠的mbd接触,将低甲基化分区(例如,无甲基化)与甲基化分区分离。珠用于从非甲基化核酸中分离出甲基化核酸。随后,顺序进行一个或更多个洗脱步骤,以洗脱具有各种甲基化水平的核酸。例如,第一组甲基化核酸可以在160mm或更高的盐浓度(例如至少150mm、至少200mm、至少300mm、至少400mm、至少500mm、至少600mm、至少700mm、至少800mm、至少900mm、至少1000mm或至少2000mm)洗脱。在这样的甲基化核酸被洗脱后,磁性分离再次用于
将较高水平的甲基化核酸与具有较低甲基化水平的核酸分离。洗脱和磁性分离步骤本身可以重复进行以产生各种分区,诸如低甲基化分区(代表无甲基化)、甲基化分区(代表低甲基化水平)和高甲基化分区(代表高甲基化水平)。
[0269]
在一些方法中,与用于亲和分离的剂结合的核酸经历洗涤步骤。洗涤步骤洗去与亲和剂弱结合的核酸。这样的核酸可以富集具有接近平均值或中值(即,在样品与剂初始接触时保持与固相结合的核酸和不与固相结合的核酸之间的中间值)程度的修饰的核酸。
[0270]
亲和分离导致至少两个且有时三个或更多个具有不同修饰程度的核酸分区。当分区仍然分开时,将至少一个分区且通常两个或三个(或更多个)分区的核酸与核酸标签连接,该核酸标签通常作为衔接子的组件提供,并且不同分区中的核酸接收将一个分区的成员与另一个分区的成员区分开的不同的标签。与同一分区的核酸分子连接的标签可以彼此相同或不同。但是如果彼此不同,则标签编码的一部分可以是共有的,以便将它们所附接的分子鉴定为属于特定分区。
[0271]
关于基于特征诸如甲基化对核酸样品进行分区的进一步细节,参见wo2018/119452,其通过引用并入本文。
[0272]
在一些实施方案中,可以基于与特定蛋白或其片段结合的核酸分子和不与该特定蛋白或其片段结合的核酸分子,将核酸分子分级为不同的分区。
[0273]
核酸分子可以基于dna-蛋白结合进行分级。蛋白-dna复合物可以基于特定的蛋白性质进行分级。这样的性质的实例包括各种表位、修饰(例如,组蛋白甲基化或乙酰化)或酶促活性。可以与dna结合并用作分级基础的蛋白的实例可以包括但不限于蛋白a和蛋白g。任何合适的方法都可以用于基于蛋白结合区将核酸分子分级。用于基于蛋白结合区将核酸分子分级的方法的实例包括但不限于sds-page、染色质免疫沉淀(chip)、肝素色谱法和非对称场流分级(asymmetrical field flow fractionation,af4)。
[0274]
在一些实施方案中,通过使核酸与甲基化结合蛋白(“mbp”)的甲基化结合结构域(“mbd”)接触来进行核酸的分区。mbd与5-甲基胞嘧啶(5mc)结合。mbd经由生物素接头与顺磁珠(诸如m-280链霉亲和素)偶联。分区为具有不同甲基化程度的级分可以通过递增的nacl浓度洗脱级分来进行。
[0275]
本文设想的mbp的实例包括,但不限于:
[0276]
(a)相比于结合未修饰的胞嘧啶,优先结合5-甲基-胞嘧啶的蛋白mecp2。
[0277]
(b)相比于结合未修饰的胞嘧啶,优先结合5-羟甲基-胞嘧啶的rpl26、prp8和dna错配修复蛋白mhs6。
[0278]
(c)相比于结合未修饰的胞嘧啶,优先结合5-甲酰基-胞嘧啶的foxki、foxk2、foxp1、foxp4和foxi3(iurlaro等人,genome biol.14:r119(2013))。
[0279]
(d)对一个或更多个甲基化核苷酸碱基特异性的抗体。
[0280]
通常,洗脱随每个分子的甲基化位点数目变化,在增加的盐浓度下,具有更多甲基化的分子洗脱。为了基于甲基化程度将dna洗脱到不同的群体中,人们可以使用一系列递增nacl浓度的洗脱缓冲液。盐浓度可以在约100nm至约2500mm nacl的范围。在实施方案中,该过程导致三(3)个分区。将分子与第一盐浓度的溶液接触,并且该溶液包含含有甲基结合结构域的分子,该分子可以与捕获部分诸如链霉亲和素附接。在第一盐浓度,一个分子群体将与mbd结合,并且一个群体将保持未结合。未结合的群体可以被分离为“低甲基化”群体。例
如,代表低甲基化形式的dna的第一分区是在低盐浓度(例如,100mm或160mm)保持未结合的分区。代表中等甲基化dna的第二分区使用中等盐浓度(例如,在100mm和2000mm之间的浓度)洗脱。这也从样品中分离。代表高甲基化形式的dna的第三分区使用高盐浓度(例如,至少约2000mm)洗脱。
[0281]
a.对分区加标签
[0282]
在一些实施方案中,对两个或更多个分区(例如,每个分区)差异性加标签。标签或索引可以是含有信息的分子,诸如核酸,该信息指示与标签缔合的分子的特征。例如,分子可以带有样品标签或样品索引(它将一个样品中的分子与不同样品中的分子区分开)、分区标签(它将一个分区中的分子与不同分区中的分子区分开)和/或分子标签/分子条形码/条形码(它将不同分子彼此区分开(在独特和非独特加标签两者的情形中))。在某些实施方案中,标签可以包括一种条形码或更多种条形码的组合。如本文使用的,术语“条形码”是指具有特定核苷酸序列的核酸分子,或指核苷酸序列本身,这取决于上下文。条形码可以具有例如10个和100个之间的核苷酸。按照特定目的期望,条形码的集合可以具有简并序列或可以有具有有一定汉明距离(hamming distance)的序列。因此,例如,分子条形码可以包括一种条形码或两种条形码(每种条形码附接到分子的不同末端)的组合。另外或可选地,对于不同的分区和/或样品,可以使用不同的分子条形码组、分子标签组或分子索引组,使得条形码通过它们的单独序列用作分子标签并且还用于基于它们是其成员的组来鉴定它们对应的分区和/或样品。
[0283]
标签可以用于标记单个多核苷酸群体分区,以便将标签(或多于一个标签)与特定分区相关联。可选地,标签可用于不采用分区步骤的本发明的实施方案中。在一些实施方案中,单一标签可用于标记特定分区。在一些实施方案中,多于一种不同的标签可以用于标记特定分区。在采用多于一种不同标签来标记特定分区的实施方案中,用于标记一个分区的标签组可以容易地与用于标记其他分区的标签组区分开。在一些实施方案中,标签可以具有另外的功能,例如标签可以用于索引样品来源或用作独特分子标识符(其可以用于通过区分测序错误与突变来改进测序数据的质量,例如,如在kinde等人,proc nat’l acad sci usa 108:9530-9535(2011);kou等人,plos one,11:e0146638(2016)中的)或用作非独特分子标识符,例如,如美国专利第9,598,731号中描述的。类似地,在一些实施方案中,标签可以具有另外的功能,例如标签可以用于索引样品来源或用作非独特分子标识符(其可以用于通过区分测序错误与突变来改进测序数据的质量)。
[0284]
在实施方案中,对分区加标签包括用分区标签对每个分区中的分子加标签。在将分区重新组合(例如,为了减少所需的测序运行次数并避免不必要的成本)和对分子测序之后,分区标签标识来源分区。在另一种实施方案中,用不同组的分子标签(例如,包含一对条形码)对不同的分区加标签。以这种方式,每个分子条形码指示来源分区,以及可用于区分分区内的分子。例如,第一组的35个条形码可以用于对第一分区中的分子加标签,而第二组的35个条形码可以用于对第二分区中的分子加标签。
[0285]
在一些实施方案中,在进行分区和用分区标签加标签之后,可以将分子汇集用于在单次运行中测序。在一些实施方案中,在例如添加分区标签和汇集之后的步骤中,将样品标签添加到分子。样品标签可以有助于将从多于一个样品产生的材料汇集用于在单次测序运行中测序。
[0286]
可选地,在一些实施方案中,分区标签可以与样品以及分区相关联。作为简单实例,第一标签可以指示第一样品的第一分区;第二标签可以指示第一样品的第二分区;第三标签可以指示第二样品的第一分区;并且第四标签可以指示第二样品的第二分区。
[0287]
虽然标签可以附接到已经基于一个或更多个特征分区的分子,但在文库中最终加标签的分子可以不再具有该特征。例如,虽然单链dna分子可能被分区和加标签,但在文库中最终加标签的分子可能是双链的。类似地,虽然dna可以经历基于不同的甲基化水平进行的分区,但在最终文库中,源自这些分子的加标签的分子可能是非甲基化的。因此,在文库中附接到分子的标签通常指示最终加标签的分子源自的“亲本分子”的特征,而不必是加标签的分子本身的特征。
[0288]
作为实例,条形码1、2、3、4等用于对第一分区中的分子加标签和标记;条形码a、b、c、d等用于对第二分区中的分子加标签和标记;并且条形码a、b、c、d等用于对第三分区中的分子加标签和标记。差异性加标签的分区可以被汇集然后测序。差异性加标签的分区可以单独测序或一起同时测序,例如,在illumina测序仪的同一流动池中。
[0289]
在测序之后,可以在各个分区的水平以及整个核酸群体水平上对读段进行分析以检测遗传变异。标签用于分选来自不同分区的读段。分析可以包括使用序列信息、基因组坐标长度、覆盖率和/或拷贝数以计算机分析来确定遗传和表观遗传变异(甲基化、染色质结构等中的一种或更多种)。在一些实施方案中,较高的覆盖率可以与基因组区域中较高的核小体占据度相关联,而较低的覆盖率可以与较低的核小体占据度或核小体缺失区(ndr)相关联。
[0290]
b.修饰的核酸分析的替代方法
[0291]
在一些实施方案中,在将核酸分区之后将衔接子添加到核酸,在其他实施方案中,可以在将核酸分区之前将衔接子添加到核酸。在一些这样的方法中,使带有不同程度修饰(例如,每个核酸分子有0、1、2、3、4、5或更多甲基基团)的核酸群体与衔接子接触,然后根据修饰的程度将群体分级。衔接子附接到群体中核酸分子的一端或两端。优选地,衔接子包含足够数目的不同标签,使得标签组合的数目导致具有相同起点和终点的两个核酸接收不同标签组合的概率高,例如95%、99%或99.9%。无论带有相同或不同标签的衔接子都可以包含相同或不同的引物结合位点,但优选地衔接子包含相同的引物结合位点。衔接子附接后,使核酸与优先结合带有修饰的核酸的剂(诸如先前描述的这样的剂)接触。将核酸分区为至少两个子样品,其不同之处在于带有修饰的核酸与剂的结合程度。例如,如果剂对带有修饰的核酸具有亲和力,则修饰被过度代表的核酸(与群体中的中位代表性相比)优先与剂结合,而修饰未被充分代表的核酸不结合剂或者更容易从剂洗脱。在分区之后,使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序,其中第一核碱基是修饰或未修饰的核碱基,第二核碱基是不同于第一核碱基的修饰或未修饰的核碱基,并且第一核碱基和第二核碱基具有相同的碱基配对特异性。然后由与衔接子内的引物结合位点结合的引物扩增核酸。在扩增之后,不同的分区然后可以并行但单独地经历进一步的处理步骤,这些步骤通常包括进一步扩增(例如,克隆扩增)和序列分析。然后可以将来自不同分区的序列数据进行比较。
[0292]
在另一种实施方案中,可以使用以下示例性程序来进行分区方案。将核酸两端连接到包含引物结合位点和标签的y型衔接子。扩增分子。然后,通过与优先结合5-甲基胞嘧
啶的抗体接触将扩增的分子分区以产生两个分区。一个分区包含缺乏甲基化的原始分子和失去甲基化的扩增拷贝。另一个分区包含具有甲基化的原始dna分子。使包含具有甲基化的原始dna分子的分区经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序,其中第一核碱基是修饰或未修饰的核碱基,第二核碱基是不同于第一核碱基的修饰或未修饰的核碱基,并且第一核碱基和第二核碱基具有相同的碱基配对特异性。然后对两个分区单独进行处理和测序,并进一步扩增甲基化分区。然后可以比较两个分区的序列数据。在该实例中,标签不是用来区分甲基化dna和未甲基化dna,而是用来区分这些分区中的不同分子,使得人们可以确定具有相同起点和终点的读段是否基于相同或不同的分子。
[0293]
本公开内容还提供了用于分析核酸群体的方法,其中至少一些核酸包含一个或更多个修饰的胞嘧啶残基,诸如5-甲基胞嘧啶和先前描述的任何其他修饰。在这些方法中,在分区之后,使核酸子样品与包含一个或更多个在5c位置处修饰的胞嘧啶残基(诸如5-甲基胞嘧啶)的衔接子接触。优选地,这样的衔接子中的所有胞嘧啶残基也都是修饰的,或者衔接子的引物结合区中所有这样的胞嘧啶都是修饰的。将衔接子附接到群体中核酸分子的两端。优选地,衔接子包含足够数目的不同标签,使得标签组合的数目导致具有相同起点和终点的两个核酸接收不同标签组合的概率高,例如95%、99%或99.9%。这样的衔接子中的引物结合位点可以是相同或不同的,但优选地是相同的。衔接子附接后,由与衔接子的引物结合位点结合的引物扩增核酸。将扩增的核酸分为第一等分试样和第二等分试样。在进行或不进行进一步处理的情况下,对第一等分试样进行序列数据测定。由此确定第一等分试样中分子的序列数据而不管核酸分子的初始甲基化状态。第二等分试样中的核酸分子经历不同地影响dna中的第一核碱基和dna中的第二核碱基的程序,其中第一核碱基包括在位置5处修饰的胞嘧啶,并且第二核碱基包括未修饰的胞嘧啶。该程序可以是亚硫酸氢盐处理或将未修饰的胞嘧啶转化为尿嘧啶的另一程序。然后将经历该程序的核酸用针对连接到核酸的衔接子的原始引物结合位点的引物扩增。现在只有最初连接到衔接子的核酸分子(不同于其扩增产物)被扩增,因为这些核酸在衔接子的引物结合位点保留了胞嘧啶,而扩增产物已失去了这些胞嘧啶残基的甲基化,这些失去甲基化的胞嘧啶残基在亚硫酸氢盐处理中已经历转化成为尿嘧啶。因此,只有群体中的原始分子(至少其中一些是甲基化的)经历扩增。扩增后,这些核酸经历序列分析。对从第一等分试样和第二等分试样确定的序列的比较尤其可以指示核酸群体中哪些胞嘧啶经历了甲基化。
[0294]
这样的分析可以使用以下示例性程序来进行。在分区后,将甲基化dna的两端连接到含有引物结合位点和标签的y形衔接子。衔接子中的胞嘧啶在位置5处被修饰(例如,5-甲基化)。衔接子的修饰用于在随后的转化步骤(例如,亚硫酸氢盐处理、tap转化或不影响修饰胞嘧啶但影响未修饰胞嘧啶的任何其他转化)中保护引物结合位点。衔接子附接后,扩增dna分子。将扩增产物分为两个等分试样用于有转化和无转化的测序。未经历转化的等分试样可以在进行或不进行进一步处理的情况下经历序列分析。另一等分试样经历不同地影响dna中的第一核碱基和dna中的第二核碱基的程序,其中第一核碱基包括在位置5处修饰的胞嘧啶,并且第二核碱基包括未修饰的胞嘧啶。该程序可以是亚硫酸氢盐处理或将未修饰的胞嘧啶转化为尿嘧啶的另一程序。当与对原始引物结合位点特异性的引物接触时,只有受胞嘧啶修饰保护的引物结合位点可以支持扩增。因此,只有原始分子而非来自第一扩增
的拷贝经历进一步扩增。进一步扩增的分子然后经历序列分析。然后可以比较来自两个等分试样的序列。如以上讨论的分离方案中的,衔接子中的核酸标签不用于区分甲基化dna和未甲基化dna,而是用于区分同一分区内的核酸分子。
[0295]
2.使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序
[0296]
本文公开的方法包括使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序的步骤,其中第一核碱基是修饰或未修饰的核碱基,第二核碱基是不同于第一核碱基的修饰或未修饰的核碱基,并且第一核碱基和第二核碱基具有相同的碱基配对特异性。在一些实施方案中,如果第一核碱基是修饰或未修饰的腺嘌呤,则第二核碱基是修饰或未修饰的腺嘌呤;如果第一核碱基是修饰或未修饰的胞嘧啶,则第二核碱基是修饰或未修饰的胞嘧啶;如果第一核碱基是修饰或未修饰的鸟嘌呤,则第二核碱基是修饰或未修饰的鸟嘌呤;如果第一核碱基是修饰或未修饰的胸腺嘧啶,则第二核碱基是修饰或未修饰的胸腺嘧啶(其中为了本步骤的目的,修饰和未修饰的尿嘧啶包括在修饰的胸腺嘧啶中)。
[0297]
在一些实施方案中,第一核碱基是修饰或未修饰的胞嘧啶,然后第二核碱基是修饰或未修饰的胞嘧啶。例如,第一核碱基可以包括未修饰的胞嘧啶(c),并且第二核碱基可以包括5-甲基胞嘧啶(mc)和5-羟甲基胞嘧啶(hmc)中的一种或更多种。可选地,第二核碱基可以包括c,并且第一核碱基可以包括mc和hmc中的一种或更多种。其它组合也是可能的,如例如在以上概述和以下讨论中指示的,诸如其中第一核碱基和第二核碱基中的一种包括mc并且另一种包括hmc。
[0298]
在一些实施方案中,不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序包括亚硫酸氢盐转化。亚硫酸氢盐处理将未修饰的胞嘧啶和某些修饰的胞嘧啶核苷酸(例如5-甲酰基胞嘧啶(fc)或5-羧基胞嘧啶(cac))转化为尿嘧啶,而其他修饰的胞嘧啶(例如,5-甲基胞嘧啶和5-羟甲基胞嘧啶)不被转化。因此,在使用亚硫酸氢盐转化的情况下,第一核碱基包括未修饰的胞嘧啶、5-甲酰基胞嘧啶、5-羧基胞嘧啶或其他受亚硫酸氢盐影响的胞嘧啶形式中的一种或更多种,并且第二核碱基可以包括mc和hmc中的一种或更多种,诸如mc和任选地hmc。对亚硫酸氢盐处理的dna的测序将读取为胞嘧啶的位置鉴定为mc位置或hmc位置。同时,读取为t的位置被鉴定为t或亚硫酸氢盐易感形式的c,诸如未修饰的胞嘧啶、5-甲酰基胞嘧啶或5-羧基胞嘧啶。因此,如本文描述的对第一子样品进行亚硫酸氢盐转化有助于使用从第一子样品获得的序列读段鉴定含有mc或hmc的位置。对于亚硫酸氢盐转化的示例性描述,参见,例如,moss等人,nat commun.2018;9:5068。在图1中说明了其中对具有高甲基化的第一子样品进行亚硫酸氢盐转化的示例性工作流程。
[0299]
在一些实施方案中,不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序包括氧化亚硫酸氢盐(ox-bs)转化。该程序首先将hmc转化为对亚硫酸氢盐易感的fc,随后进行亚硫酸氢盐转化。因此,当使用氧化亚硫酸氢盐转化时,第一核碱基包括未修饰的胞嘧啶、fc、cac、hmc或其他受亚硫酸氢盐影响的胞嘧啶形式中的一种或更多种,并且第二核碱基包括mc。对ox-bs转化的dna的测序将读取为胞嘧啶的位置鉴定为mc位置。同时,读取为t的位置被鉴定为t、hmc或亚硫酸氢盐易感形式的c,诸如未修饰的胞嘧啶、fc或hmc。因此,如本文描述的对第一子样品进行ox-bs转化有助于使用从第一子样品获得的
序列读段鉴定含有mc的位置。对于氧化亚硫酸氢盐转化的示例性描述,参见,例如,booth等人,science 2012;336:934-937。
[0300]
在一些实施方案中,不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序包括tet辅助的亚硫酸氢盐(tab)转化。在tab转化中,hmc被保护不被转化,并且mc在亚硫酸氢盐处理之前被氧化,因此最初由mc占据的位置被转化为u,而最初由hmc占据的位置保持为胞嘧啶的受保护形式。例如,如yu等人,cell 2012;149:1368-80中描述的,β-葡糖基转移酶可以用于保护hmc(形成5-葡糖基羟甲基胞嘧啶(ghmc)),然后tet蛋白诸如mtet1可以用于将mc转化为cac,并且然后亚硫酸氢盐处理可以用于将c和cac转化为u,而ghmc保持不受影响。因此,当使用tab转化时,第一核碱基包括未修饰的胞嘧啶、fc、cac、mc或其他受亚硫酸氢盐影响的胞嘧啶形式中的一种或更多种,并且第二核碱基包括hmc。对tab转化的dna的测序将读取为胞嘧啶的位置鉴定为hmc位置。同时,读取为t的位置被识别为t、mc或亚硫酸氢盐易感形式的c,诸如未修饰的胞嘧啶、fc或cac。因此,如本文描述的对第一子样品进行tab转化有助于使用从第一子样品获得的序列读段鉴定含有hmc的位置。
[0301]
在一些实施方案中,不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序包括tet辅助的取代硼烷还原剂转化,任选地其中取代硼烷还原剂是2-甲基吡啶硼烷、吡啶硼烷、叔丁基胺硼烷或氨硼烷。在tet辅助的取代硼烷还原剂转化中,使用tet蛋白将mc和hmc转化为cac,而不影响未修饰的c。然后,通过用2-甲基吡啶硼烷(pic-硼烷)或其他取代硼烷还原剂(诸如吡啶硼烷、叔丁基胺硼烷或氨硼烷)处理,将cac和fc(如果存在的话)转化为二氢尿嘧啶(dhu),也不影响未修饰的c。参见,例如,liu等人,nature biotechnology 2019;37:424
–
429(例如,在补充图1和补充说明7)。在测序中dhu被读取为t。因此,当使用这种类型的转化时,第一核碱基包括mc、fc、cac或hmc中的一种或更多种,并且第二核碱基包括未修饰的胞嘧啶。对转化的dna的测序将读取为胞嘧啶的位置鉴定为未修饰的c位置。同时,读取为t的位置被鉴定为t、mc、fc、cac或hmc。因此,如本文描述的对第一子样品进行tap转化有助于使用从第一子样品获得的序列读段鉴定含有未修饰的c的位置。该程序包括tet-辅助的吡啶硼烷测序(taps),在以下中进一步详细描述:liu等人2019,同上。
[0302]
可选地,对hmc的保护(例如,使用βgt)可以与tet辅助的取代硼烷还原剂转化组合。hmc可以如以上提到的通过使用βgt的葡糖基化形成ghmc得到保护。用tet蛋白诸如mtet1处理,然后将mc转化为cac但不转化c或ghmc。然后通过用pic-硼烷或其他取代硼烷还原剂(诸如吡啶硼烷、叔丁基胺硼烷或氨硼烷)处理将cac转化为dhu,也不影响未修饰的c或ghmc。因此,当使用tet辅助的取代硼烷还原剂转化时,第一核碱基包括mc,并且第二核碱基包括未修饰胞嘧啶或hmc中的一种或更多种,诸如未修饰的胞嘧啶和任选地hmc、fc和/或cac。对转化的dna的测序将读取为胞嘧啶的位置鉴定为hmc位置或未修饰的c位置。同时,读取为t的位置被鉴定为t、fc、cac或mc。因此,如本文描述的对第一子样品进行tapsβ转化有助于使用从第一子样品获得的序列读段在一方面区分含有未修饰的c或hmc的位置与含有mc的位置。对于这种类型的转化的示例性描述,参见,例如,liu等人,nature biotechnology 2019;37:424
–
429。
[0303]
在一些实施方案中,不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序包括化学辅助的取代硼烷还原剂转化,任选地其中取代硼烷还原剂是2-甲基
吡啶硼烷、吡啶硼烷、叔丁基胺硼烷或氨硼烷。在化学辅助的取代硼烷还原剂转化中,氧化剂诸如高钌酸钾(kruo4)(也适用于ox-bs转化)用于特异性地将hmc氧化为fc。用pic-硼烷或其他取代硼烷还原剂(诸如吡啶硼烷、叔丁基胺硼烷或氨硼烷)处理将fc和cac转化为dhu,但不影响mc或未修饰的c。因此,当使用这种类型的转化时,第一核碱基包括hmc、fc和cac中的一种或更多种,并且第二核碱基包括未修饰的胞嘧啶或mc中的一种或更多种,诸如未修饰的胞嘧啶和任选地mc。对转化的dna的测序将读取为胞嘧啶的位置鉴定为mc位置或未修饰的c位置。同时,读取为t的位置被鉴定为t、fc、cac或hmc。因此,如本文描述的对第一子样品进行这种类型的转化有助于使用从第一子样品获得的序列读段在一方面区分含有未修饰的c或mc的位置与含有hmc的位置。对于这种类型的转化的示例性描述,参见,例如,liu等人,nature biotechnology 2019;37:424
–
429。
[0304]
在一些实施方案中,不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序包括apobec偶联的表观遗传(ace)转化。在ace转化中,aid/apobec家族dna脱氨基酶诸如apobec3a(a3a)用于使未修饰的胞嘧啶和mc脱氨基,而不使hmc、fc或cac脱氨基。因此,当使用ace转化时,第一核碱基包括未修饰的c和/或mc(例如,未修饰的c和任选地mc),并且第二核碱基包括hmc。对ace转化的dna的测序将读取为胞嘧啶的位置鉴定为hmc位置、fc位置或cac位置。同时,读取为t的位置被鉴定为t、未修饰的c或mc。因此,如本文描述的对第一子样品进行ace转化有助于使用从第一子样品获得的序列读段来区分含有hmc的位置与含有mc或未修饰的c的位置。对于ace转化的示例性描述,参见,例如,schutsky等人,nature biotechnology 2018;36:1083
–
1090。
[0305]
在一些实施方案中,不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序包括对第一核碱基的酶促转化,例如,如em-seq中的。参见,例如,vaisvila r,等人(2019)em-seq:detection of dna methylation at single base resolution from picograms of dna.biorxiv;doi:10.1101/2019.12.20.884692,在www.biorxiv.org/content/10.1101/2019.12.20.884692v1可获得。例如,tet2和t4-βgt可用于将5mc和5hmc转化为不能被脱氨基酶(例如,apobec3a)脱氨基的底物,并且然后脱氨基酶(例如,apobec3a)可用于使未修饰的胞嘧啶脱氨基,将其转化为尿嘧啶。
[0306]
在一些实施方案中,不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的方法包括将最初包含第一核碱基的dna与最初不包含第一核碱基的dna分离。在一些这样的实施方案中,第一核碱基是hmc。可以使用包括将最初包含第一核碱基的位置生物素化的标记程序将最初包含第一核碱基的dna与其他dna分离。在一些实施方案中,首先将第一核碱基用含叠氮基的部分(诸如含有葡糖基-叠氮基的部分)衍生化。然后,含叠氮基的部分可以用作附接生物素的试剂,例如通过huisgen环加成化学。然后,可以使用生物素结合剂,诸如亲和素、中性亲和素(neutravidin)(具有约6.3的等电点的去糖基化亲和素)或链霉亲和素,将最初包含第一核碱基现在被生物素化的dna与最初不包含第一核碱基的dna分离。用于将最初包含第一核碱基的dna与最初不包含第一核碱基的dna分离的方法的实例是hmc-seal,hmc-seal标记hmc以形成β-6-叠氮基-葡糖基-5-羟甲基胞嘧啶,并且然后通过huisgen环加成附接生物素部分,随后使用生物素结合剂来分离生物素化dna与其他dna。对于hmc-seal的示例性描述,参见,例如,han等人,mol.cell 2016;63:711-719。这种方法可用于鉴定包含一个或更多个hmc核碱基的片段。
[0307]
在一些实施方案中,在这样的分离之后,该方法还包括对最初包含第一核碱基的dna、最初不包含第一核碱基的dna和第二子样品的dna中的每一个差异性加标签。该方法还可以包括在差异性加标签之后汇集最初包含第一核碱基的dna、最初不包含第一核碱基的dna和第二子样品的dna。然后,可以在同一测序池中对最初包含第一核碱基的dna、最初不包含第一核碱基的dna和第二子样品的dna进行测序,同时保持使用差异性标签来分辨特定读段是来自最初包含第一核碱基的dna、最初不包含第一核碱基的dna还是第二子样品的dna的分子的能力。
[0308]
在一些实施方案中,第一核碱基是修饰或未修饰的腺嘌呤,并且第二核碱基是修饰或未修饰的腺嘌呤。在一些实施方案中,修饰的腺嘌呤是n
6-甲基腺嘌呤(ma)。在一些实施方案中,修饰的腺嘌呤是n
6-甲基腺嘌呤(ma)、n
6-羟甲基腺嘌呤(hma)或n
6-甲酰基腺嘌呤(fa)中的一种或更多种。
[0309]
包括甲基化dna免疫沉淀(medip)的技术可以用于分离含有修饰的碱基(诸如ma)的dna与其他dna。参见,例如,kumar等人,frontiers genet.2018;9:640;greer等人,cell 2015;161:868-878。对ma特异性的抗体在sun等人,bioessays 2015;37:1155-62中描述。针对各种修饰的核碱基(诸如胸腺嘧啶/尿嘧啶的形式,包括卤化形式,诸如5-溴尿嘧啶)的抗体是商业上可获得的。各种修饰的碱基也可以基于它们的碱基配对特异性的改变来检测。例如,次黄嘌呤是腺嘌呤的修饰形式,其可以由脱氨基产生并且在测序中被读取为g。参见,例如,us专利第8,486,630号;brown,genomes,第2版,john wiley&sons,inc.,new york,n.y.,2002,第14章,“mutation,repair,and recombination.”[0310]
3.富集/捕获步骤;扩增;衔接子;条形码
[0311]
在一些实施方案中,本文公开的方法包括捕获dna诸如cfdna的一个或更多个靶区组的步骤。可以使用本领域中已知的任何合适的方法来进行捕获。
[0312]
在一些实施方案中,捕获包括使待捕获的dna与靶特异性探针组接触。靶特异性探针组可以具有本文描述的靶特异性探针组的任何特征,包括但不限于上文阐述的实施方案和下文与探针有关的章节中的特征。可以对本文公开的方法过程中制备的一个或更多个子样品进行捕获。在一些实施方案中,从至少第一子样品或第二子样品,例如至少第一子样品和第二子样品中捕获dna。在第一子样品经历分离步骤(例如,分离最初包含第一核碱基(例如,hmc)的dna与最初不包含第一核碱基的dna,诸如hmc-seal)的情况下,可以对最初包含第一核碱基(例如,hmc)的dna、最初不包含第一核碱基的dna和第二子样品中的任何一个、任何两个或全部进行捕获。在一些实施方案中,对子样品进行差异性加标签(例如,如本文描述的),并且然后在进行捕获之前进行汇集。
[0313]
捕获步骤可以使用适于特定核酸杂交的条件进行,该条件通常在某种程度上取决于探针的特征,诸如长度、碱基组成等。鉴于本领域有关核酸杂交的一般知识,本领域技术人员对适当的条件将是熟悉的。在一些实施方案中,形成靶特异性探针和dna的复合物。
[0314]
在一些实施方案中,本文描述的方法包括捕获从测试受试者获得的cfdna的多于一个靶区组。靶区包括表观遗传靶区,所述表观遗传靶区可以显示出甲基化水平和/或片段化模式的差异,这取决于它们来源于肿瘤细胞还是来源于健康细胞。靶区还包括序列可变靶区,所述序列可变靶区可以显示出序列差异,这取决于它们来源于肿瘤细胞还是来源于健康细胞。捕获步骤产生cfdna分子的捕获组,并且在cfdna分子的捕获组中,对应于序列可
变靶区组的cfdna分子以比对应于表观遗传靶区组的cfdna分子更大的捕获产量被捕获。对于捕获步骤、捕获产量和相关方面的另外的讨论,参见wo2020/160414,为了所有目的将其通过引用并入本文。
[0315]
在一些实施方案中,本文描述的方法包括使从测试受试者获得的cfdna与靶特异性探针组接触,其中靶特异性探针组被配置为以比对应于表观遗传靶区组的cfdna更大的捕获产量捕获对应于序列可变靶区组的cfdna。
[0316]
以比对应于表观遗传靶区组的cfdna更大的捕获产量捕获对应于序列可变靶区组的cfdna是有益的,因为以足够的置信度或准确度分析序列可变靶区可能需要的测序深度比分析表观遗传靶区可能需要的测序深度更大。确定片段化模式(例如,测试转录起始位点或ctcf结合位点的扰动)或片段丰度(例如,在高甲基化分区和低甲基化分区中)所需的数据量通常小于确定癌症相关序列突变的存在或不存在所需的数据量。以不同的产量捕获靶区组可以有助于在同一测序运行中(例如,使用汇集的混合物和/或在同一测序池中)将靶区测序到不同的测序深度。
[0317]
在各种实施方案中,该方法还包括将捕获的cfdna测序到,例如,对表观遗传靶区组和序列可变靶区组不同程度的测序深度,与本文讨论的一致。
[0318]
在一些实施方案中,将靶特异性探针和dna的复合物与未结合到靶特异性探针的dna分离。例如,在靶特异性探针共价地或非共价地结合到固体支持物的情况下,可以使用洗涤或抽吸步骤来分离未结合的材料。可选地,在复合物具有不同于未结合材料的色谱特性的情况下(例如,在探针包含结合色谱树脂的配体的情况下),可以使用色谱法。
[0319]
如本文其他处详细讨论的,靶特异性探针组可以包括多于一个组,诸如针对序列可变靶区组的探针和针对表观遗传靶区组的探针。在一些这样的实施方案中,在同一容器中同时使用针对序列可变靶区的探针和针对表观遗传靶区的探针进行捕获步骤,例如,针对序列可变靶区组和表观遗传靶区组的探针在同一组合物中。该方法提供了相对效率更高的工作流程。在一些实施方案中,针对序列可变靶区组的探针的浓度大于针对表观遗传靶区组的探针的浓度。
[0320]
可选地,在第一容器中用序列可变靶区探针组并在第二容器中用表观遗传靶区探针组进行捕获步骤,或者在第一时间和第一容器用序列可变靶区探针组并在第一时间之前或之后的第二时间用表观遗传靶区探针组进行接触步骤。该方法允许制备单独的第一组合物和第二组合物,所述第一组合物和第二组合物包含对应于序列可变靶区组的捕获的dna和对应于表观遗传靶区组的捕获的dna。所述组合物可以按期望单独地处理(例如,基于甲基化进行分级,如本文其他处描述的),并以适当比例重新组合以提供用于进一步处理和分析诸如测序的材料。
[0321]
在一些实施方案中,扩增dna。在一些实施方案中,在捕获步骤之前进行扩增。在一些实施方案中,在捕获步骤之后进行扩增。
[0322]
在一些实施方案中,dna中包含衔接子。这可以与扩增程序同时进行,例如,通过在引物的5’部分中提供衔接子,例如,如以上描述的。可选地,衔接子可以通过其他方法诸如连接添加。
[0323]
在一些实施方案中,dna中包含标签,标签可以是条形码或包含条形码。标签可以有助于鉴定核酸的来源。例如,条形码可用于允许在汇集多于一个样品用于并行测序之后
鉴定dna来自的来源,例如,受试者。这可以与扩增程序同时进行,例如,通过在引物的5’部分中提供条形码,例如,如以上描述的。在一些实施方案中,衔接子和标签/条形码由同一引物或引物组提供。例如,条形码可以位于衔接子的3’和引物的靶杂交部分的5’。可选地,条形码可以通过其他方法添加,诸如连接,任选地与衔接子一起在同一连接底物中。
[0324]
关于扩增、标签和条形码的另外的细节在以下“方法的一般特征”章节中讨论,这些细节可以在可行的程度上与任前述实施方案和“引言及概述”章节中阐述的实施方案组合。
[0325]
4.捕获组
[0326]
在一些实施方案中,提供dna(例如,cfdna)的捕获组。对于所公开的方法,dna的捕获组可以例如通过在如本文描述的分区步骤之后进行捕获步骤来提供。捕获组可以包括对应于序列可变靶区组的dna、对应于表观遗传靶区组的dna或其组合。在一些实施方案中,当针对靶区的尺寸(足迹尺寸)的差异进行归一化时,捕获的序列可变靶区dna的量大于捕获的表观遗传靶区dna的量。
[0327]
可选地,可以提供分别包括对应于序列可变靶区组的dna和对应于表观遗传靶区组的dna的第一捕获组和第二捕获组。可以组合第一捕获组和第二捕获组以提供组合的捕获组。
[0328]
在一些其中包括对应于序列可变靶区组和表观遗传靶区组的dna的捕获组包括如以上讨论的组合的捕获组的实施方案中,对应于序列可变靶区组的dna可以以比对应于表观遗传靶区组的dna更大的浓度(例如,1.1倍至1.2倍大的浓度、1.2倍至1.4倍大的浓度、1.4倍至1.6倍大的浓度、1.6倍至1.8倍大的浓度、1.8倍至2.0倍大的浓度、2.0倍至2.2倍大的浓度、2.2倍至2.4倍大的浓度、2.4倍至2.6倍大的浓度、2.6倍至2.8倍大的浓度、2.8倍至3.0倍大的浓度、3.0倍至3.5倍大的浓度、3.5倍至4.0、4.0倍至4.5倍大的浓度、4.5倍至5.0倍大的浓度、5.0倍至5.5倍大的浓度、5.5倍至6.0倍大的浓度、6.0倍至6.5倍大的浓度、6.5倍至7.0倍大、7.0倍至7.5倍大的浓度、7.5倍至8.0倍大的浓度、8.0倍至8.5倍大的浓度、8.5倍至9.0倍大的浓度、9.0倍至9.5倍大的浓度、9.5倍至10.0倍大的浓度、10倍至11倍大的浓度、11倍至12倍大的浓度、12倍至13倍大的浓度、13倍至14倍大的浓度、14倍至15倍大的浓度、15倍至16倍大的浓度、16倍至17倍大的浓度、17倍至18倍大的浓度、18倍至19倍大的浓度、19倍至20倍大的浓度、20倍至30倍大的浓度、30倍至40倍大的浓度、40倍至50倍大的浓度、50倍至60倍大的浓度、60倍至70倍大的浓度、70倍至80倍大的浓度、80倍至90倍大的浓度、90倍至100倍大的浓度、10倍至20倍大的浓度、10倍至40倍大的浓度、10倍至50倍大的浓度、10倍至70倍大的浓度或10倍至100倍大的浓度)存在。浓度差异的程度按针对靶区足迹尺寸的归一化计算,如定义章节中讨论的。
[0329]
a.表观遗传靶区组
[0330]
表观遗传靶区组可以包括一个或更多个类型的靶区,所述靶区可以区分来自赘生性(例如,肿瘤或癌症)细胞的dna与来自健康细胞(例如,非赘生性循环细胞)的dna。本文详细讨论了这样的区域的示例性类型。表观遗传靶区组还可以包括一个或更多个对照区,例如,如本文描述的。
[0331]
在一些实施方案中,表观遗传靶区组具有至少100kb(例如至少200kb、至少300kb或至少400kb)的足迹。在一些实施方案中,表观遗传靶区组具有100kb-1000kb(例如,
100kb-200kb、200kb-300kb、300kb-400kb、400kb-500kb、500kb-600kb、600kb-700kb、700kb-800kb、800kb-900kb和900kb-1,000kb)范围内的足迹。
[0332]
i.高甲基化可变靶区
[0333]
在一些实施方案中,表观遗传靶区组包括一个或更多个高甲基化可变靶区。通常,高甲基化可变靶区是指这样的区域,在该区域中,例如在cfdna样品中,所观察到的甲基化水平的增加指示样品(例如cfdna)含有由赘生性细胞(诸如肿瘤细胞或癌细胞)产生的dna的可能性增加。例如,肿瘤抑制基因启动子的高甲基化已经被重复观察到。参见,例如,kang等人,genome biol.18:53(2017)及其中引用的参考文献。在实例中,高甲基化可变靶区可以包括这样的区域,相对于来自相同类型的健康组织的dna,癌性组织中该区域的甲基化不一定不同,但相对于健康受试者中典型的cfdna,该区域的甲基化确实不同(例如,具有更多甲基化)。例如,当癌症的存在导致细胞死亡(诸如对应于癌症的组织类型的细胞凋亡)增加时,可以至少部分地使用这样的高甲基化可变靶区来检测这样的癌症。在一些实施方案中,高甲基化可变靶区包括一个或更多个基因组区域,其中在癌症受试者中这些区域中的cfdna分子的甲基化状态相对于来自健康受试者的cfdna没有差异,但是在这些区域中高甲基化cfdna的存在/增加的量指示特定的组织类型(例如,癌症来源),并且由于增加的凋亡(例如,肿瘤脱落)进入循环中作为cfdna呈现。
[0334]
对结肠直肠癌中甲基化可变靶区的广泛讨论在lam等人,biochim biophys acta.1866:106-20(2016)中提供。这些包括vim、sept9、itga4、osm4、gata4和ndrg4。在表1中提供了基于结肠直肠癌(crc)研究的示例性高甲基化可变靶区组。这些基因中的许多种可能与结肠直肠癌之外的癌症有关;例如,tp53被广泛认为是至关重要的肿瘤抑制因子,并且该基因的基于高甲基化的失活可能是常见的致癌机制。
[0335]
表1.基于crc研究的示例性高甲基化靶区。
res.61:4556
–
4560,(2001)。
[0339]
在表2中提供了基于肺癌研究的示例性高甲基化可变靶区组。这些基因中的许多种可能与肺癌之外的癌症有关;例如,casp8(胱天蛋白酶8)是程序性细胞死亡的关键酶,并且该基因的基于高甲基化的失活可能是常见的致癌机制,不限于肺癌。另外,一些基因在表1和表2两者中都出现,指示一般性。
[0340]
表2.基于肺癌研究的示例性高甲基化可变靶区
[0341][0342][0343]
涉及表2中鉴定的靶区的任何前述实施方案可以与涉及表1中鉴定的靶区的任何前述实施方案组合。在一些实施方案中,高甲基化可变靶区包括表1或表2中列出的多于一个基因座,例如,表1或表2中列出的基因座中的至少10%、20%、30%、40%、50%、60%、70%、80%、90%或100%。
[0344]
另外的高甲基化靶区可以例如从癌症基因组图谱(the cancer genome atlas)获得。kang等人,genome biology 18:53(2017)描述了使用来自乳腺、结肠、肾、肝和肺的高甲
基化靶区构建称为癌症定位器(cancerlocator)的概率方法。在一些实施方案中,高甲基化靶区可以是对一个或更多个类型的癌症特异性的。因此,在一些实施方案中,高甲基化靶区包括一个、两个、三个、四个或五个高甲基化靶区子集,所述高甲基化靶区子集集体地显示出乳腺癌、结肠癌、肾癌、肝癌和肺癌中的一种、两种、三种、四种或五种中的高甲基化。
[0345]
ii.低甲基化可变靶区
[0346]
全面低甲基化是在多种癌症中普遍观察到的现象。参见,例如,hon等人,genome res.22:246-258(2012)(乳腺癌);ehrlich,epigenomics 1:239-259(2009)(提到对结肠癌、卵巢癌、前列腺癌、白血病、肝细胞癌和宫颈癌中低甲基化的观察的综述文章)。例如,在健康细胞中正常被甲基化的区域(诸如重复元件(例如,line1元件、alu元件、着丝粒串联重复序列、着丝粒周围串联重复序列和卫星dna)和基因间区域)在肿瘤细胞中可能显示出减少的甲基化。因此,在一些实施方案中,表观遗传靶区组包括低甲基化可变靶区,其中所观察到的甲基化水平的降低指示样品(例如,cfdna)含有由赘生性细胞(诸如肿瘤细胞或癌细胞)产生的dna的可能性增加。在实例中,低甲基化可变靶区可以包括这样的区域,相对于来自相同类型的健康组织的dna,癌性组织中该区域的甲基化状态不一定不同,但相对于健康受试者中典型的cfdna,该区域的甲基化确实不同(例如,甲基化较少)。例如,当癌症的存在导致细胞死亡(诸如对应于癌症的组织类型的细胞凋亡)增加时,可以至少部分地使用这样的低甲基化可变靶区来检测这样的癌症。在一些实施方案中,低甲基化可变靶区包括一个或更多个基因组区域,其中在癌症受试者中这些区域中的cfdna分子的甲基化状态相对于来自健康受试者的cfdna没有差异,但是在这些区域中低甲基化cfdna的存在/增加的量指示特定的组织类型(例如,癌症来源),并且以伴随增加的凋亡(例如,肿瘤脱落)进入循环中的cfdna呈现。
[0347]
在一些实施方案中,低甲基化可变靶区包括重复元件和/或基因间区域。在一些实施方案中,重复元件包括line1元件、alu元件、着丝粒串联重复序列、着丝粒周围串联重复序列和/或卫星dna中的一种、两种、三种、四种或五种。
[0348]
显示出癌症相关的低甲基化的示例性特定基因组区域包括人类1号染色体的核苷酸8403565-8953708和151104701-151106035。在一些实施方案中,低甲基化可变靶区与这些区域中的一个或两个重叠或者包括这些区域中的一个或两个。
[0349]
iii.ctcf结合区
[0350]
ctcf是有助于染色质组织并经常与黏连蛋白共定位的dna结合蛋白。ctcf结合位点的扰动已在许多不同的癌症中被报道。参见,例如,katainen等人,nature genetics,doi:10.1038/ng.3335,2015年6月8日在线出版;guo等人,nat.commun.9:1520(2018)。ctcf结合导致cfdna中可识别的模式,其可以通过测序检测,例如通过片段长度分析。关于基于测序的片段长度分析的细节在snyder等人,cell 164:57-68(2016);wo 2018/009723和us20170211143a1中提供,它们中的每一项通过引用并入本文。
[0351]
因此,ctcf结合的扰动导致cfdna片段化模式的变异。因此,ctcf结合位点代表片段化可变靶区的类型。
[0352]
有许多已知的ctcf结合位点。参见,例如,ctcfbsdb(ctcf结合位点数据库),在互联网上在insulatordb.uthsc.edu/可获得;cuddapah等人,genome res.19:24-32(2009);martin等人,nat.struct.mol.biol.18:708-14(2011);rhee等人,cell.147:1408-19
(2011),它们中的每一项通过引用并入。示例性ctcf结合位点位于8号染色体上的核苷酸56014955-56016161处和13号染色体上的核苷酸95359169-95360473处。
[0353]
因此,在一些实施方案中,表观遗传靶区组包括ctcf结合区。在一些实施方案中,ctcf结合区包括至少10个、20个、50个、100个、200个或500个ctcf结合区、或者10-20个、20-50个、50-100个、100-200个、200-500个或500-1000个ctcf结合区,例如,诸如上文描述的ctcf结合区,或者ctcfbsdb或上文引用的文章cuddapah等人、martin等人或rhee等人中的一项或更多项中的ctcf结合区。
[0354]
在一些实施方案中,至少一些ctcf位点可以是甲基化或未甲基化的,其中甲基化状态与细胞是否是癌细胞相关。在一些实施方案中,表观遗传靶区组包括ctcf结合位点的上游和下游区域至少100bp、至少200bp、至少300bp、至少400bp、至少500bp、至少750bp、至少1000bp。
[0355]
iv.转录起始位点
[0356]
赘生性细胞中转录起始位点也可以显示出扰动。例如,在造血谱系的健康细胞中各转录起始位点处的核小体组织(其在健康个体中对cfdna有实质性贡献)可以不同于赘生性细胞中这些转录起始位点处的核小体组织。这导致了不同的cfdna模式,可以通过测序来检测,如在snyder等人,cell 164:57-68(2016);wo 2018/009723和us20170211143a1中普遍讨论的。在另一实例中,癌性组织中转录起始位点相对于来自同一类型的健康组织的dna在表观遗传上不一定不同,但相对于健康受试者中典型的cfdna在表观遗传上(例如,对于核小体组织)确实不同。例如,当癌症的存在导致细胞死亡(诸如对应于癌症的组织类型的细胞凋亡)增加时,可以至少部分地使用这样的转录起始位点来检测这样的癌症。
[0357]
因此,转录起始位点的扰动也导致cfdna片段化模式的变异。因此,转录起始位点也代表片段化可变靶区的类型。
[0358]
人类转录起始位点从dbtss(人类转录起始位点数据库)可获得,在互联网上在dbtss.hgc.jp可获得,并在in yamashita等人,nucleic acids res.34(数据库期号):d86
–
d89(2006)中描述,其通过引用并入本文。
[0359]
因此,在一些实施方案中,表观遗传靶区组包括转录起始位点。在一些实施方案中,转录起始位点包括至少10个、20个、50个、100个、200个或500个转录起始位点,或10-20个、20-50个、50-100个、100-200个、200-500个或500-1000个转录起始位点,例如,诸如dbtss中列出的转录起始位点。在一些实施方案中,至少一些转录起始位点可以是甲基化或未甲基化的,其中甲基化状态与细胞是否是癌细胞相关。在一些实施方案中,表观遗传靶区组包括转录起始位点的上游和下游区域至少100bp、至少200bp、至少300bp、至少400bp、至少500bp、至少750bp、至少1000bp。
[0360]
v.聚焦扩增
[0361]
尽管聚焦扩增是体细胞突变,但它们可以通过基于读段频率的测序以类似于检测某些表观遗传改变诸如甲基化改变的方法的方式来检测。因此,在癌症中可以显示出聚焦扩增的区域可以被包括在表观遗传靶区组中,并且可以包括ar、braf、ccnd1、ccnd2、ccne1、cdk4、cdk6、egfr、erbb2、fgfr1、fgfr2、kit、kras、met、myc、pdgfra、pik3ca和raf1中的一种或更多种。例如,在一些实施方案中,表观遗传靶区组包括前述靶中的至少2种、3种、4种、5种、6种、7种、8种、9种、10种、11种、12种、13种、14种、15种、16种、17种或18种。
[0362]
vi.甲基化对照区
[0363]
纳入对照区来帮助数据验证可能是有用的。在一些实施方案中,表观遗传靶区组包括对照区,所述对照区预期在基本上所有样品中都是甲基化或未甲基化的,不管dna源自癌细胞还是正常细胞。在一些实施方案中,表观遗传靶区组包括预期在基本上所有样品中都是低甲基化的对照低甲基化区。在一些实施方案中,表观遗传靶区组包括预期在基本上所有样品中都是高甲基化的对照高甲基化区。
[0364]
b.序列可变靶区组
[0365]
在一些实施方案中,序列可变靶区组包括已知在癌症中经历体细胞突变的多于一个区域。
[0366]
在一些方面,序列可变靶区组靶向所选择的多于一个不同的基因或基因组区域(“组(panel)”),使得确定比例的患有癌症的受试者在组中的一个或多于一个不同的基因或基因组区域中表现出遗传变异或肿瘤标志物。可以将组选择为将用于测序的区域限定为固定数目的碱基对。可以将组选择为对期望的量的dna进行测序,例如,通过调节探针的亲和力和/或量,如本文其他处描述的。还可以将组选择为实现期望的序列读段深度。可以将组选择为对一定量的测序的碱基对实现期望的序列读段深度或序列读段覆盖率。可以将组选择为对检测样品中一种或更多种遗传变异实现理论灵敏度、理论特异性和/或理论准确度。
[0367]
用于检测该组区域的探针可以包括用于检测感兴趣的基因组区域(热点区域)的探针以及核小体感知探针(例如,kras密码子12和13),并且可以设计成基于分析cfdna覆盖率和受核小体结合模式影响的片段尺寸变异和gc序列组成来优化捕获。本文使用的区域还可以包括基于核小体位置和gc模式优化的非热点区域。
[0368]
感兴趣的基因组位置列表的实例可见于表3和表4。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表3中的至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个或70个基因的至少一部分。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表3中的至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个或70个snv。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表3中的至少1个、至少2个、至少3个、至少4个、至少5个或6个融合。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表3中的至少1个、至少2个或3个插入/缺失中的至少一部分。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表4中的至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个、至少70个或73个基因的至少一部分。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表4中的至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个、至少70个或73个snv。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表4中的至少1个、至少2个、至少3个、至少4个、至少5个或6个融合。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表4中的至少1个、至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、
至少14个、至少15个、至少16个、至少17个或18个插入/缺失中的至少一部分。这些感兴趣的基因组位置中的每一个可以被鉴定为特定组的骨架区域或热点区域。感兴趣的热点基因组位置列表的实例可见于表5。在一些实施方案中,本公开内容的方法中使用的序列可变靶区组包含表5中的至少1个、至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、至少14个、至少15个、至少16个、至少17个、至少18个、至少19个或至少20个基因的至少一部分。每个热点基因组区域都列出了几个特征,包括相关基因、其所在的染色体、基因组的代表基因的基因座的起始和终止位置、以碱基对计的基因座长度、基因覆盖的外显子和感兴趣的特定基因组区域可以寻求捕获的关键特征(例如,突变类型)。
[0369]
表3
[0370][0371]
表4
[0372][0373]
表5
[0374]
[0375][0376]
另外或可选地,从文献中可获得合适的靶区组。例如,gale等人,plos one 13:e0194630(2018),其通过引用并入本文,描述了一组35个癌症相关基因靶,可用作序列可变靶区组的一部分或全部。这35个靶是akt1、alk、braf、ccnd1、cdk2a、ctnnb1、egfr、erbb2、esr1、fgfr1、fgfr2、fgfr3、foxl2、gata3、gna11、gnaq、gnas、hras、idh1、idh2、kit、kras、med12、met、myc、nfe2l2、nras、pdgfra、pik3ca、ppp2r1a、pten、ret、stk11、tp53和u2af1。
[0377]
在一些实施方案中,序列可变靶区组包含来自至少10个、20个、30个或35个癌症相关基因,诸如以上列出的癌症相关基因的靶区。
[0378]
5.受试者
[0379]
在一些实施方案中,dna(例如,cfdna)从患有癌症的受试者获得。在一些实施方案中,dna(例如,cfdna)从怀疑患有癌症的受试者获得。在一些实施方案中,dna(例如,cfdna)从患有肿瘤的受试者获得。在一些实施方案中,dna(例如,cfdna)从怀疑患有肿瘤的受试者获得。在一些实施方案中,dna(例如,cfdna)从患有赘生物的受试者获得。在一些实施方案中,dna(例如,cfdna)从怀疑患有赘生物的受试者获得。在一些实施方案中,dna(例如,cfdna)从处于从肿瘤、癌症或赘生物缓解(例如,在化学疗法、手术切除、放射或其组合之后)的受试者获得。在任一前述实施方案中,癌症、肿瘤或赘生物或者疑似的癌症、肿瘤或赘生物可以是肺、结肠、直肠、肾、乳腺、前列腺或肝的。在一些实施方案中,癌症、肿瘤或赘生物或者疑似的癌症、肿瘤或赘生物是肺的。在一些实施方案中,癌症、肿瘤或赘生物或者疑似的癌症、肿瘤或赘生物是结肠的或直肠的。在一些实施方案中,癌症、肿瘤或赘生物或者疑似的癌症、肿瘤或赘生物是乳腺的。在一些实施方案中,癌症、肿瘤或赘生物或者疑似的癌症、肿瘤或赘生物是前列腺的。在任一前述实施方案中,受试者可以是人类受试者。
[0380]
6.测序
[0381]
通常,侧翼为衔接子的样品核酸可以在预先扩增或不预先扩增的情况下进行测序。测序方法包括,例如,sanger测序、高通量测序、焦磷酸测序、合成测序、单分子测序、纳米孔测序、半导体测序、连接测序、杂交测序、数字基因表达(helicos)、下一代测序(ngs)、单分子合成测序(smss)(helicos)、大规模并行测序、克隆单分子阵列(solexa)、鸟枪测序、ion torrent、oxford纳米孔、roche genia、maxim-gilbert测序、引物步移(primer walking)和使用pacbio、solid、ion torrent或纳米孔平台的测序。测序反应可以在各种样品处理单元中进行,样品处理单元可以是基本上同时处理多于一个样品组的多道、多通道、多孔或其他装置。样品处理单元还可以包括多于一个样品室以能够同时处理多于一个运行。
[0382]
可以对一种或更多种形式的核酸进行测序反应,其中至少一种形式的核酸已知含有癌症或其他疾病的标志物。也可以对样品中存在的任何核酸片段进行测序反应。在一些实施方案中,对基因组的测序覆盖率可以小于5%、10%、15%、20%、25%、30%、40%、50%、60%、70%、80%、90%、95%、99%、99.9%或100%。在一些实施方案中,测序反应可以提供对基因组的至少5%、10%、15%、20%、25%、30%、40%、50%、60%、70%或80%的测序覆盖率。测序覆盖可以对至少5个、10个、20个、70个、100个、200个或500个不同的基因进行,或者对最多5000个、2500个、1000个、500个或100个不同的基因进行。
[0383]
同时测序反应可以使用多重测序进行。在一些情况下,可以用至少1000个、2000个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10000个、50000个、100,000个测序反应对无细胞核酸测序。在其他情况下,可以用少于1000个、2000个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10000个、50000个、100,000个测序反应对无细胞核酸测序。测序反应可以依次进行或同时进行。可以对所有或部分测序反应进行后续数据分析。在一些情况下,可以对至少1000个、2000个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10000个、50000个、100,000个测序反应进行数据分析。在其他情况下,可以对少于1000个、2000个、3000个、4000个、5000个、6000个、7000个、8000个、9000个、10000个、50000个、100,000个测序反应进行数据分析。示例性读段深度是1000-50000个读段/基因座(碱基)。
[0384]
a.差异性测序深度
[0385]
在一些实施方案中,将对应于序列可变靶区组的核酸测序到比对应于表观遗传靶区组的核酸更大的测序深度。例如,对应于序列变异靶区组的核酸的测序深度可以是对应于表观遗传靶区组的核酸的测序深度的至少1.25倍、1.5倍、1.75倍、2倍、2.25倍、2.5倍、2.75倍、3倍、3.5倍、4倍、4.5倍、5倍、6倍、7倍、8倍、9倍、10倍、11倍、12倍、13倍、14倍、或15倍大、或者1.25倍至1.5倍、1.5倍至1.75倍、1.75倍至2倍、2倍至2.25倍、2.25倍至2.5倍、2.5倍至2.75倍、2.75倍至3倍、3倍至3.5倍、3.5倍至4倍、4倍至4.5倍、4.5倍至5倍、5倍至5.5倍、5.5倍至6倍、6倍至7倍、7倍至8倍、8倍至9倍、9倍至10倍、10倍至11倍、11倍至12倍、13倍至14倍、14倍至15倍或15倍至100倍大。在一些实施方案中,所述测序深度是至少2倍大。在一些实施方案中,所述测序深度是至少5倍大。在一些实施方案中,所述测序深度是至少10倍大。在一些实施方案中,所述测序深度是4倍至10倍大。在一些实施方案中,所述测序深度是4倍至100倍大。这些实施方案中的每一种都涉及将对应于序列可变靶区组的核酸测序到比对应于表观遗传靶区组的核酸更大的测序深度的程度。
[0386]
在一些实施方案中,对应于序列可变靶区组的捕获的cfdna和对应于表观遗传靶区组的捕获的cfdna被同时测序,例如在同一测序池(诸如illumina测序仪的流动池)中和/或在同一组合物中;所述同一组合物可以是由重新组合单独捕获的组产生的汇集的组合物,或者通过在同一容器中捕获对应于序列可变靶区组的cfdna和对应于表观遗传靶区组的捕获的cfdna而获得的组合物。
[0387]
7.分析
[0388]
在一些实施方案中,本文描述的方法包括鉴定由肿瘤(或赘生性细胞或癌细胞)产生的dna的存在或不存在。
[0389]
本方法可以用于诊断受试者中状况特别是癌症的存在或不存在,以表征状况(例如,对癌症进行分期或确定癌症的异质性),监测状况对治疗的响应,实现对状况发展或状况后续进程的风险的预后。本公开内容也可以用于确定特定治疗选择的效力。如果治疗是成功的,则更多的癌症可能死亡并且使dna脱落,所以成功的治疗选择可增加受试者的血液中检测到的拷贝数变异或罕见突变的量。在其他实例中,这可能不会发生。在另一实例中,也许某些治疗选择可能与癌症随时间推移的遗传谱相关。这种相关性可以用于选择疗法。
[0390]
另外,如果观察到癌症在治疗之后处于缓解中,本方法可以用于监测残留疾病或疾病的复发。
[0391]
可以检测到的类型和数目的癌症可以包括血癌、脑癌、肺癌、皮肤癌、鼻癌、喉癌、肝癌、骨癌、淋巴瘤、胰腺癌、皮肤癌、肠癌、直肠癌、甲状腺癌、膀胱癌、肾癌、口腔癌、胃癌、固态瘤、异质性肿瘤、同质性肿瘤等。癌症的类型和/或分期可以根据遗传变异检测,包括突变、罕见突变、插入/缺失、拷贝数变异、颠换、易位、倒位、缺失、非整倍性、部分非整倍性、多倍性、染色体不稳定性、染色体结构改变、基因融合、染色体融合、基因截短、基因扩增、基因复制、染色体损伤、dna损伤、核酸化学修饰的异常改变、表观遗传模式的异常改变和核酸5-甲基胞嘧啶的异常改变。
[0392]
遗传数据还可以用于表征特定形式的癌症。癌症在组成和分期两方面通常是异质性的。遗传谱数据可以允许表征癌症的具体亚型,该表征在该具体亚型的诊断或治疗中可能是重要的。该信息还可以为受试者或从业者提供关于具体类型癌症的预后的线索,并且
允许受试者或从业者根据疾病的进展调整治疗选择。一些癌症可以进展而变得更具侵袭性和遗传不稳定性。其他癌症可以保持良性的、非活动的、或休眠的。本公开内容的系统和方法可以用于确定疾病进展。
[0393]
此外,本公开内容的方法可以用于表征受试者的异常状况的异质性。这样的方法可以包括,例如生成源自受试者的细胞外多核苷酸的遗传谱,其中所述遗传谱包括由拷贝数变异和罕见突变分析得到的多于一个数据。在一些实施方案中,异常状况是癌症。在一些实施方案中,异常状况可以是导致异质性基因组群体的状况。在癌症的实例中,已知一些肿瘤包含处于癌症的不同分期的肿瘤细胞。在其他实例中,异质性可以包括疾病的多个病灶。再次,在癌症的实例中,可以存在多个肿瘤病灶,或许其中一个或更多个病灶为已从原发部位扩散的转移的结果。
[0394]
本方法可以用于生成或剖析源自异质性疾病中不同细胞的遗传信息的总和的指纹图谱或数据集。该数据集可以包括单独的或组合的拷贝数变异、表观遗传变异和突变分析。
[0395]
本方法可以用于诊断、预后、监测或观察癌症或其他疾病。在一些实施方案中,本文的方法不涉及诊断、预后或监测胎儿,并因此不涉及非侵入性产前测试。在其他实施方案中,这些方法可以用于妊娠受试者以诊断、预后、监测或观察未出生受试者的癌症或其他疾病,所述未出生受试者的dna和其他多核苷酸可以与母体分子共循环。
[0396]
用于通过ngs(所述ngs包括使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序的步骤)对mbd-珠分区的文库进行分子标签鉴定的示例性方法如下:
[0397]
1.使用甲基结合结构域蛋白-珠纯化试剂盒对提取的dna样品(例如,从人类样品提取的血浆dna,其已任选地经历了如本文描述的靶捕获)进行物理分区,保留来自过程的所有洗脱物用于下游处理。
[0398]
2.将差异性分子标签和ngs可行性衔接子(ngs-enabling adapter)序列并行应用于每个分区。例如,将高甲基化分区、残留甲基化(
‘
洗涤’)分区和低甲基化分区与带有分子标签的ngs衔接子连接。
[0399]
3.使高甲基化分区经历不同地影响dna中的第一核碱基和dna中的第二核碱基的程序,诸如本文描述的那些程序中的任一种。
[0400]
4.重新组合所有加分子标签的分区,并使用衔接子特异性dna引物序列进行随后扩增。
[0401]
5.对重新组合和扩增的总文库进行捕获/杂交,靶向感兴趣的基因组区域(例如,癌症特异性遗传变异和差异性甲基化区域)。
[0402]
6.重新扩增捕获的dna文库,附加样品标签。将不同的样品汇集,并在ngs仪器上进行多重测定。
[0403]
7.对ngs数据进行生物信息学分析,其中使用分子标签来鉴定独特分子,以及将样品去卷积为差异性mbd分区的分子。该分析可以与标准的基因测序/变异检测同时产生基因组区域的相对5-甲基胞嘧啶的信息。
[0404]
在本文描述的方法(包括但不限于以上示出的方法)的一些实施方案中,分子标签由不被不同地影响dna中的第一核碱基和dna中的第二核碱基的程序所改变的核苷酸组成,
诸如本文描述的那些核苷酸中的任一种(例如,当所述程序是亚硫酸氢盐转化或不影响mc的任何其他转化时为mc以及a、t和g;当所述程序是不影响hmc的转化时为hmc以及a、t和g;等等)。在本文描述的方法(包括但不限于以上显示出的方法)的一些实施方案中,分子标签不包含被不同地影响dna中的第一核碱基和dna中的第二核碱基的程序所改变的核苷酸,诸如本文描述的那些核苷酸中的任一种(例如,当所述程序是亚硫酸氢盐转化或影响c的任何其他转化时,标签不包含未修饰的c;当所述程序是影响mc的转化时,标签不包含mc;当所述程序是影响hmc的转化时,标签不包含hmc;等等)。
[0405]
通常,不同地影响dna中的第一核碱基和dna中的第二核碱基的程序可以替代地在将差异性分子标签和ngs可行性衔接子序列并行应用于每个分区的步骤之前进行。例如,这可以在不同地影响dna中的第一核碱基和dna中的第二核碱基的程序是分离的情况下进行,诸如hmc-seal,并且在这样的情况下,分离的群体本身可以相对于彼此差异性加标签。这样的示例性方法如下:
[0406]
1.使用甲基结合结构域蛋白-珠纯化试剂盒对提取的dna样品(例如,从人类样品提取的血浆dna,其已任选地经历了如本文描述的靶捕获)进行物理分区,保留来自过程的所有洗脱物用于下游处理。
[0407]
2.使高甲基化分区经历不同地影响dna中的第一核碱基和dna中的第二核碱基的程序,诸如本文描述的那些程序中的任一种。
[0408]
3.将差异性分子标签和ngs可行性衔接子序列并行应用于每个分区。例如,将高甲基化分区(或在适用的情况下,高甲基化分区的两个或更多个子分区)、残留甲基化(
‘
洗涤’)分区和低甲基化分区与带有分子标签的ngs衔接子连接。
[0409]
4.重新组合所有加分子标签的分区,并使用衔接子特异性dna引物序列进行随后扩增。
[0410]
5.对重新组合和扩增的总文库进行捕获/杂交,靶向感兴趣的基因组区域(例如,癌症特异性遗传变异和差异性甲基化区域)。
[0411]
6.重新扩增捕获的dna文库,附加样品标签。将不同的样品汇集,并在ngs仪器上进行多重测定。
[0412]
7.对ngs数据进行生物信息学分析,其中使用分子标签来鉴定独特分子,以及将样品去卷积为差异性mbd分区的分子。该分析可以与标准的基因测序/变异检测同时产生基因组区域的相对5-甲基胞嘧啶的信息。
[0413]
8.示例性工作流程
[0414]
本文提供了用于分区和文库制备的示例性工作流程。在一些实施方案中,分区和文库制备工作流程的一些或所有特征可以组合使用。
[0415]
a.分区
[0416]
在一些实施方案中,将样品dna(例如,在5ng至200ng之间)与甲基结合结构域(mbd)缓冲液和与mbd蛋白缀合的磁珠混合并孵育过夜。在该孵育期间甲基化dna(高甲基化dna)结合磁珠上的mbd蛋白。用含递增盐浓度的缓冲液将非甲基化(低甲基化dna)或甲基化较少(中等甲基化)的dna从珠上洗下。例如,可以从这样的洗涤中获得含有非甲基化dna、低甲基化dna和/或中等甲基化dna的一个、两个或更多个级分。最后,使用高盐缓冲液将重度甲基化的dna(高甲基化dna)从mbd蛋白洗脱。在一些实施方案中,这些洗涤产生具有递增的
甲基化水平的dna的三个分区(低甲基化分区、中等甲基化级分和高甲基化分区)。
[0417]
在一些实施方案中,将dna的三个分区脱盐并浓缩以准备用于文库制备的酶促步骤。
[0418]
b.文库制备
[0419]
在一些实施方案中(例如,在将分区中的dna浓缩之后),使分区的dna可连接,例如,通过延伸dna分子的末端突出端,并将腺苷残基添加到片段的3’末端,并使每个dna片段的5’末端磷酸化。添加dna连接酶和衔接子以将每个分区的dna分子的每个末端与衔接子连接。这些衔接子含有与用于其他分区的衔接子中的分区标签可区分的分区标签(例如,非随机、非独特条形码)。在使分区的dna可连接并进行连接之前或之后,使高甲基化分区经历不同地影响dna中的第一核碱基和dna中的第二核碱基的程序,诸如本文描述的程序中的任一种。在不同地影响dna中的第一核碱基和dna中的第二核碱基的程序将高甲基化分区进一步分区的情况下,应在该程序之后进行衔接子的连接,以便可以对高甲基化分区的子分区差异性加标签。然后,将三个(或更多)分区汇集在一起并进行扩增(例如,通过pcr,诸如使用对衔接子特异性的引物)。
[0420]
pcr之后,扩增的dna可以进行清洗并在富集之前浓缩。使扩增的dna与本文描述的靶向感兴趣的特定区域的探针集合(其可以是,例如生物素化的rna探针)接触。孵育混合物,例如过夜,例如在盐缓冲液中。捕获探针(例如,使用链霉亲和素磁珠)并将其与未捕获的扩增dna分离,诸如通过一系列盐洗涤,从而富集样品。富集之后,通过pcr扩增富集的样品。在一些实施方案中,pcr引物含有样品标签,从而将样品标签掺入到dna分子中。在一些实施方案中,将来自不同样品的dna汇集在一起,并且然后进行多重测序,例如使用illumina novaseq测序仪。
[0421]
c.某些公开的方法的另外的特征
[0422]
1.样品
[0423]
样品可以是从受试者分离的任何生物样品。样品可以是身体样品。样品可以包括身体组织,诸如已知或怀疑的实体瘤、全血、血小板、血清、血浆、粪便、红细胞、白血细胞(white blood cell)或白细胞(leucocyte)、内皮细胞、活检组织、脑脊液、滑液、淋巴液、腹水、间质液或细胞外液,细胞之间的间隙中的流体(包括龈沟液)、骨髓、胸腔积液、脑脊液、唾液、粘液、痰、精液、汗液、尿液。样品优选为体液,特别是血液及其级分,以及尿液。样品可以是最初从受试者中分离出来的形式,或者可以进行进一步处理以去除或添加组分,诸如细胞,或者相对于其他组分对一种组分进行富集。因此,用于分析的优选的体液是含有无细胞核酸的血浆或血清。可以从受试者分离或获得样品,并将其运送到样品分析地点。样品可以在合适的温度,例如室温、4℃、-20℃和/或-80℃保存和运输。可以在样品分析地点从受试者分离或获得样品。受试者可以是人类、哺乳动物、动物、伴侣动物、服务动物或宠物。受试者可以患有癌症。受试者可以没有癌症或可检测到的癌症症状。受试者可以已经用一种或更多种癌症疗法治疗过,例如,化学疗法、抗体、疫苗或生物制剂中的任一种或更多种。受试者可以处于缓解中。受试者可以被诊断或可以未被诊断为对癌症或任何癌症相关的基因突变/紊乱易感。在一些实施方案中,样品是从肿瘤组织活检中获得的多核苷酸样品。
[0424]
血浆的体积可以取决于对测序的区域期望的读段深度。示例性体积为0.4ml-40ml、5ml-20ml、10ml-20ml。例如,体积可以是0.5ml、1ml、5ml、10ml、20ml、30ml或40ml。取
样的血浆体积可以是5ml至20ml。
[0425]
样品可以包含各种量的包含基因组等同物的核酸。例如,约30ng dna的样品可以含有约10,000(104)个单倍体人类基因组等同物,并且在cfdna的情况下,可以含有约2000亿(2x10
11
)个个体多核苷酸分子。类似地,约100ng的dna的样品可以包含约30,000个单倍体人类基因组等同物,并且在cfdna的情况下,可以包含约6000亿个个体分子。
[0426]
样品可以包含来自不同来源的核酸,例如,来自同一受试者的细胞和无细胞的核酸,来自不同受试者的细胞和无细胞的核酸。样品可以包含携带突变的核酸。例如,样品可以包含携带种系突变和/或体细胞突变的dna。种系突变是指存在于受试者的种系dna中的突变。体细胞突变是指来源于受试者的体细胞例如癌细胞的突变。样品可以包含携带癌症相关突变(例如,癌症相关体细胞突变)的dna。样品可以包含表观遗传变异(即化学或蛋白修饰),其中表观遗传变异与遗传变异(诸如癌症相关突变)的存在相关。在一些实施方案中,样品包含与遗传变异的存在相关的表观遗传变异,其中样品不包含所述遗传变异。
[0427]
扩增前样品中无细胞核酸的示例性量的范围为约1fg至约1μg,例如1pg至200ng、1ng至100ng、10ng至1000ng。例如,量可以是上至约600ng、上至约500ng、上至约400ng、上至约300ng、上至约200ng、上至约100ng、上至约50ng或上至约20ng的无细胞核酸分子。量可以是至少1fg、至少10fg、至少100fg、至少1pg、至少10pg、至少100pg、至少1ng、至少10ng、至少100ng、至少150ng或至少200ng的无细胞核酸分子。量可以是上至1飞克(fg)、10fg、100fg、1皮克(pg)、10pg、100pg、1ng、10ng、100ng、150ng或200ng的无细胞核酸分子。方法可以包括从样品中获得1飞克(fg)至200ng的无细胞核酸分子。
[0428]
无细胞核酸是不包含在细胞内或以其他方式与细胞结合的核酸,或者换言之,在去除完整细胞后保留在样品中的核酸。无细胞核酸包括dna、rna及其杂合体(hybrid),包括基因组dna、线粒体dna、sirna、mirna、循环rna(crna)、trna、rrna、小核仁rna(snorna)、piwi-相互作用rna(pirna)、长非编码rna(长ncrna)或这些中的任一种的片段。无细胞核酸可以是双链的、单链的或其杂合体。无细胞核酸可以通过分泌或细胞死亡过程例如细胞坏死和凋亡释放到体液中。一些无细胞核酸是从癌细胞释放到体液中的,例如循环肿瘤dna(ctdna)。其他无细胞核酸是从健康细胞释放的。在一些实施方案中,cfdna是无细胞胎儿dna(cffdna)。在一些实施方案中,无细胞核酸由肿瘤细胞产生。在一些实施方案中,无细胞核酸由肿瘤细胞和非肿瘤细胞的混合物产生。
[0429]
无细胞核酸具有约100-500个核苷酸的示例性尺寸分布,其中110个至约230个核苷酸的分子代表约90%的分子,具有约168个核苷酸的众数,并且第二个小峰在240个至440个核苷酸之间的范围内。
[0430]
无细胞核酸可以通过分级或分区步骤从体液分离,在所述分级或分区步骤中,如存在于溶液中的无细胞核酸与体液中的完整细胞和其他不可溶组分分离。分区可以包括诸如离心或过滤的技术。可选地,体液中的细胞可以被裂解,并且无细胞核酸和细胞核酸一起被处理。通常,在添加缓冲液和洗涤步骤之后,核酸可以用醇沉淀。可以使用进一步的清洗步骤诸如基于二氧化硅的柱以去除污染物或盐。可以在整个反应中添加非特异性批量载体核酸(bulk carrier nucleic acid),诸如c1dna、dna或蛋白,用于亚硫酸氢盐测序、杂交和/或连接,以优化程序的某些方面,诸如产量。
[0431]
在这样的处理后,样品可以包括各种形式的核酸,包括双链dna、单链dna和单链
rna。在一些实施方案中,单链dna和rna可以被转化成双链形式,因此它们被包括在后续处理和分析步骤中。
[0432]
样品中的双链dna分子和转化为双链dna分子的单链核酸分子可以在一端或两端连接到衔接子。通常,在所有四种标准核苷酸存在的情况下,通过用具有5
’‑3’
聚合酶和3
’‑5’
外切核酸酶(或校对功能)的聚合酶处理使双链分子平末端化。klenow大片段和t4聚合酶是合适的聚合酶的实例。平末端的dna分子可以与至少部分地双链的衔接子(例如,y形或钟形衔接子)连接。可选地,可以将互补核苷酸添加到样品核酸和衔接子的平末端以促进连接。本文设想了平末端连接和粘性末端连接两者。在平末端连接中,核酸分子和衔接子标签两者都具有平末端。在粘性末端连接中,通常核酸分子带有“a”突出端并且衔接子带有“t”突出端。
[0433]
2.扩增
[0434]
侧翼为衔接子的样品核酸可以通过pcr和其他扩增方法来扩增。扩增通常是由结合到侧翼是待扩增的dna分子的衔接子中的引物结合位点的引物引发。扩增方法可以包括由热循环产生的变性、退火和延伸的循环,或可以是如转录介导的扩增中的等温循环。其他扩增方法包括连接酶链式反应、链置换扩增、基于核酸序列的扩增和基于自我维持序列(self-sustained sequence)的复制。
[0435]
在一些实施方案中,本方法用t-尾和c-尾衔接子进行dsdna连接,这导致至少50%、60%、70%或80%的双链核酸的扩增。优选地,相对于单独用t尾衔接子进行的对照方法,本方法使扩增分子的量或数目增加至少10%、15%或20%。
[0436]
3.标签
[0437]
包括条形码的标签可以掺入衔接子中或以其他方式连接到衔接子。标签可以通过连接、重叠延伸pcr等方法掺入。
[0438]
a.分子加标签策略
[0439]
在一些实施方案中,核酸分子(来自多核苷酸样品)可以用样品索引和/或分子条形码(通常称为“标签”)进行加标签。标签可以通过化学合成、连接反应(例如,平末端连接或粘性末端连接)或重叠延伸聚合酶链式反应(pcr)等方法掺入衔接子中或以其他方式连接到衔接子。这样的衔接子可以最终连接到靶核酸分子。在其他实施方案中,通常应用一轮或更多轮扩增循环(例如,pcr扩增)以使用常规核酸扩增方法将样品索引引入核酸分子。扩增可以在一种或更多种反应混合物(例如,阵列中的多于一个微孔)中进行。分子条形码和/或样品索引可以同时或以任何顺序引入。在一些实施方案中,在进行序列捕获步骤之前和/或之后引入分子条形码和/或样品索引。在一些实施方案中,在探针捕获之前仅引入分子条形码,并且在进行序列捕获步骤之后引入样品索引。在一些实施方案中,在进行基于探针的捕获步骤之前,引入分子条形码和样品索引两者。在一些实施方案中,在进行序列捕获步骤之后引入样品索引。在一些实施方案中,分子条形码经由连接(例如,平末端连接或粘性末端连接)通过衔接子掺入样品中的核酸分子(例如,cfdna分子)。在一些实施方案中,通过重叠延伸聚合酶链式反应(pcr),将样品索引掺入样品中的核酸分子(例如,cfdna分子)。典型地,序列捕获方案包括引入与靶核酸序列(例如基因组区域的编码序列,并且这样的区域的突变与癌症类型相关)互补的单链核酸分子。
[0440]
在一些实施方案中,标签可以位于样品核酸分子的一端或两端。在一些实施方案
中,标签是预定序列的或随机序列的或半随机序列的寡核苷酸。在一些实施方案中,标签的长度可以小于约500个、200个、100个、50个、20个、10个、9个、8个、7个、6个、5个、4个、3个、2个或1个核苷酸。标签可以随机或非随机地连接到样品核酸。
[0441]
在一些实施方案中,每个样品用样品索引或样品索引的组合独特地加标签。在一些实施方案中,样品或子样品的每个核酸分子用分子条形码或分子条形码的组合独特地加标签。在其他实施方案中,可以使用多于一个分子条形码,使得分子条形码不必在多于一个中彼此独特(例如,非独特分子条形码)。在这些实施方案中,分子条形码通常附接(例如,通过连接)至个体分子,使得分子条形码和可以与其附接的序列的组合产生可以被单独地追溯的独特序列。与内源性序列信息(例如,对应于样品中原始核酸分子序列的开始(起始)和/或结束(终止)基因组位置(location)/位置(position),对应于样品中原始核酸分子序列的起始和终止基因组位置,映射到参考序列的序列读段的开始(起始)和/或结束(终止)基因组位置(location)/位置(position),映射到参考序列的序列读段的起始和终止基因组位置,序列读段在一端或两端的子序列,序列读段的长度和/或样品中原始核酸分子的长度)组合检测非独特分子条形码通常允许将独特身份指定至特定分子。在一些实施方案中,开始区域包括测序读段与参考序列对齐的5’末端处的前1个、前2个、前5个、前10个、前15个、前20个、前25个、前30个或至少前30个碱基位置。在一些实施方案中,结束区域包括测序读段与参考序列对齐的3’末端处的最后1个、最后2个、最后5个、最后10个、最后15个、最后20个、最后25个、最后30个或至少最后30个碱基位置。个体序列读段的长度或碱基对数目也任选地用于将独特身份指定至特定分子。如本文描述的,来自已经指定了独特身份的核酸单链的片段从而可以允许后续鉴定来自该亲本链和/或互补链的片段。
[0442]
在某些实施方案中,用于独特地鉴定一个类别中数目(z)的分子的不同标签的数目可以在2*z、3*z、4*z、5*z、6*z、7*z、8*z、9*z、10*z、11*z、12*z、13*z、14*z、15*z、16*z、17*z、18*z、19*z、20*z或100*z中的任一个(例如,下限)和100,000*z、10,000*z、1000*z或100*z(例如,上限)中的任一个之间。在一些实施方案中,分子条形码以一组标识符(例如,独特或非独特分子条形码的组合)的预期比率引入到样品中的分子。一种示例形式使用连接到靶分子两端的约2个至约1,000,000个不同的分子条形码序列、或约5个至约150个不同的分子条形码序列、或约20个至约50个不同的分子条形码序列。可选地,可以使用约25个至约1,000,000个不同的分子条形码序列。例如,可以使用20-50
×
20-50个分子条形码序列(即,20-50个不同的分子条形码序列之一可以附接到靶分子的每一端)。这样数目的标识符通常足以使具有相同起点和终点的不同分子具有接收不同标识符组合的高可能性(例如,至少94%、99.5%、99.99%或99.999%)。在一些实施方案中,约80%、约90%、约95%或约99%的分子具有相同的分子条形码组合。
[0443]
在一些实施方案中,使用在例如美国专利申请第20010053519号、第20030152490号和第20110160078号以及美国专利第6,582,908号、第7,537,898号、第9,598,731号和第9,902,992号中所述的方法和系统来进行反应中独特或非独特分子条形码的分配,所述专利申请和专利中的每一个通过引用以其整体特此并入。可选地,在一些实施方案中,可以仅使用内源序列信息(例如,起始和/或终止位置、序列一端或两端的子序列和/或长度)来鉴定样品的不同核酸分子。
[0444]
4.诱饵组;捕获部分
[0445]
如以上描述的,可以使样品中的核酸经历捕获步骤,其中捕获具有靶序列的分子用于后续分析。靶捕获可以包括使用诱饵组,所述诱饵组包括用捕获部分(诸如,生物素或以下提到的其他实例)标记的寡核苷酸诱饵。探针可以具有被选择为平铺(tile)跨越一组区域(诸如基因)的序列。在一些实施方案中,诱饵组可以对靶区组(诸如序列可变靶区组和表观遗传靶区的靶区组)分别具有较高和较低的捕获产量,如本文其他处讨论的。在允许靶分子与诱饵杂交的条件下,将这些诱饵组与样品组合。然后,使用捕获部分来分离捕获的分子。例如,基于珠的链霉亲和素的生物素捕获部分。这样的方法在例如2017年12月26日颁布的美国专利9,850,523中进一步描述,该专利通过引用并入本文。
[0446]
捕获部分包含但不限于生物素、亲和素、链霉亲和素、包含特定核苷酸序列的核酸、被抗体识别的半抗原和磁性可吸引颗粒。提取部分可以是结合对的成员,诸如生物素/链霉亲和素或半抗原/抗体。在一些实施方案中,附接到分析物的捕获部分被它的结合对捕获,该结合对附接到可分离部分,诸如磁性可吸引颗粒或可以通过离心沉淀的大颗粒。捕获部分可以是允许将带有捕获部分的核酸与缺乏捕获部分的核酸亲和分离的任何类型的分子。示例性捕获部分是生物素或寡核苷酸,所述生物素允许通过与连接或可连接到固相的链霉亲和素结合而进行亲和分离,所述寡核苷酸允许通过与连接或可连接到固相的互补寡核苷酸结合而进行亲和分离。
[0447]
d.靶特异性探针的集合
[0448]
在一些实施方案中,在本文描述的方法中使用靶特异性探针的集合。在一些实施方案中,靶特异性探针的集合包括对序列可变靶区组特异性的靶结合探针和对表观遗传靶区组特异性的靶结合探针。在一些实施方案中,对序列可变靶区组特异性的靶结合探针的捕获产量比对表观遗传靶区组特异性的靶结合探针的捕获产量高(例如,高至少2倍)。在一些实施方案中,靶特异性探针的集合被配置为具有比其对表观遗传靶区特异性的捕获产量高(例如,高至少2倍)的对序列可变靶区特异性的捕获产量。
[0449]
在一些实施方案中,对序列可变靶区组特异性的靶结合探针的捕获产量比对表观遗传靶区组特异性的靶结合探针的捕获产量高至少1.25倍、1.5倍、1.75倍、2倍、2.25倍、2.5倍、2.75倍、3倍、3.5倍、4倍、4.5倍、5倍、6倍、7倍、8倍、9倍、10倍、11倍、12倍、13倍、14倍或15倍。在一些实施方案中,对序列可变靶区组特异性的靶结合探针的捕获产量比对表观遗传靶区组特异性的靶结合探针的捕获产量高1.25倍至1.5倍、1.5倍至1.75倍、1.75倍至2倍、2倍至2.25倍、2.25倍至2.5倍、2.5倍至2.75倍、2.75倍至3倍、3倍至3.5倍、3.5倍至4倍、4倍至4.5倍、4.5倍至5倍、5倍至5.5倍、5.5倍至6倍、6倍至7倍、7倍至8倍、8倍至9倍、9倍至10倍、10倍至11倍、11倍至12倍、13倍至14倍或14倍至15倍。
[0450]
在一些实施方案中,靶特异性探针的集合被配置为具有比其对表观遗传靶区组的捕获产量高至少1.25倍、1.5倍、1.75倍、2倍、2.25倍、2.5倍、2.75倍、3倍、3.5倍、4倍、4.5倍、5倍、6倍、7倍、8倍、9倍、10倍、11倍、12倍、13倍、14倍或15倍的对序列可变靶区组特异性的捕获产量。在一些实施方案中,靶特异性探针的集合被配置为具有比其对表观遗传靶区组特异性的捕获产量高1.25倍至1.5倍、1.5倍至1.75倍、1.75倍至2倍、2倍至2.25倍、2.25倍至2.5倍、2.5倍至2.75倍、2.75倍至3倍、3倍至3.5倍、3.5倍至4倍、4倍至4.5倍、4.5倍至5倍、5倍至5.5倍、5.5倍至6倍、6倍至7倍、7倍至8倍、8倍至9倍、9倍至10倍、10倍至11倍、11倍至12倍、13倍至14倍或14倍至15倍的对序列可变靶区组特异性的捕获产量。
[0451]
探针的集合可以被配置为以各种方式(包括浓度、不同的长度和/或化学(例如,影响亲和力的化学)及其组合)提供对序列可变靶区组的更高的捕获产量。亲和力可以通过调节探针长度和/或包括核苷酸修饰来调节,如以下讨论的。
[0452]
在一些实施方案中,对序列可变靶区组特异性的靶特异性探针以比对表观遗传靶区组特异性的靶特异性探针更高的浓度存在。在一些实施方案中,对序列可变靶区组特异性的靶结合探针的浓度比对表观遗传靶区组特异性的靶结合探针的浓度高至少1.25倍、1.5倍、1.75倍、2倍、2.25倍、2.5倍、2.75倍、3倍、3.5倍、4倍、4.5倍、5倍、6倍、7倍、8倍、9倍、10倍、11倍、12倍、13倍、14倍或15倍。在一些实施方案中,对序列可变靶区组特异性的靶结合探针的浓度比对表观遗传靶区组特异性的靶结合探针的浓度高1.25倍至1.5倍、1.5倍至1.75倍、1.75倍至2倍、2倍至2.25倍、2.25倍至2.5倍、2.5倍至2.75倍、2.75倍至3倍、3倍至3.5倍、3.5倍至4倍、4倍至4.5倍、4.5倍至5倍、5倍至5.5倍、5.5倍至6倍、6倍至7倍、7倍至8倍、8倍至9倍、9倍至10倍、10倍至11倍、11倍至12倍、13倍至14倍或14倍至15倍。在这样的实施方案中,浓度可以指每组中单独探针的平均质量/体积浓度。
[0453]
在一些实施方案中,与对表观遗传靶区组特异性的靶特异性探针相比,对序列可变靶区组特异性的靶特异性探针对其靶具有更高的亲和力。亲和力可以以本领域技术人员已知的任何方式调节,包括通过使用不同的探针化学。例如,某些核苷酸修饰,诸如胞嘧啶5-甲基化(在某些序列的情况下)、在2’糖位置提供杂原子的修饰、和lna核苷酸,可以增加双链核酸的稳定性,这意味着具有这样的修饰的寡核苷酸对其互补序列具有相对更高的亲和力。参见,例如,severin等人,nucleic acids res.39:8740
–
8751(2011);freier等人,nucleic acids res.25:4429
–
4443(1997);美国专利第9,738,894号。此外,较长的序列长度通常将提供增加的亲和力。其他核苷酸修饰,诸如核碱基次黄嘌呤对鸟嘌呤的取代,通过减少寡核苷酸与其互补序列之间的氢键的量来减少亲和力。在一些实施方案中,对序列可变靶区组特异性的靶特异性探针具有增加其对其靶的亲和力的修饰。在一些实施方案中,可选地或另外地,对表观遗传靶区组特异性的靶特异性探针具有降低其对其靶的亲和力的修饰。在一些实施方案中,对序列可变靶区组特异性的靶特异性探针比对表观遗传靶区组特异性的靶特异性探针具有更长的平均长度和/或更高的平均解链温度。这些实施方案可以相互组合和/或具有如以上讨论的浓度差异,以实现捕获产量的期望的倍数差异,诸如以上描述的任何倍数差异或其范围。
[0454]
在一些实施方案中,靶特异性探针包含捕获部分。捕获部分可以是本文描述的任何捕获部分,例如生物素。在一些实施方案中,将靶特异性探针连接到固体支持物,例如,共价地或非共价地,诸如通过捕获部分的结合对的相互作用。在一些实施方案中,固体支持物是珠,诸如磁珠。
[0455]
在一些实施方案中,对序列可变靶区组特异性的靶特异性探针和/或对表观遗传靶区组特异性的靶特异性探针是如以上讨论的诱饵组,例如,包含捕获部分和被选择为平铺跨越一组区域(诸如基因)的序列的探针。
[0456]
在一些实施方案中,靶特异性探针以单一组合物提供。单一组合物可以是溶液(液体或冷冻的)。可选地,单一组合物可以是冻干物。
[0457]
可选地,靶特异性探针可以作为多于一种组合物提供,例如,包括第一组合物和第二组合物,所述第一组合物包含对表观遗传靶区组特异性的探针,所述第二组合物包含对
序列可变靶区组特异性的探针。这些探针可以以适当的比例混合,以提供在浓度和/或捕获产量上具有任何前述倍数差异的组合的探针组合物。可选地,它们可以在单独的捕获程序中使用(例如,用于样品的等分试样或依次用于同一样品),以提供分别包含捕获的表观遗传靶区和捕获的序列可变靶区的第一组合物和第二组合物。
[0458]
1.对表观遗传靶区特异性的探针
[0459]
针对表观遗传靶区组的探针可以包括对一个或更多个类型的靶区特异性的探针,所述靶区能够区分来自赘生性(例如,肿瘤或癌症)细胞与来自健康细胞(例如,非赘生性循环细胞)的dna。在本文(例如,在以上关于捕获组的章节中)详细讨论了这样的区域的示例性类型。针对表观遗传靶区组的探针还可以包括针对一个或更多个对照区的探针,例如,如本文描述的。
[0460]
在一些实施方案中,表观遗传靶区探针组的探针具有至少100kb(例如,至少200kb、至少300kb或至少400kb)的足迹。在一些实施方案中,针对表观遗传靶区组的探针具有100-1000kb(例如,100-200kb、200-300kb、300-400kb、400-500kb、500-600kb、600-700kb、700-800kb、800-900kb和900-1000kb)范围内的足迹。在一些实施方案中,表观遗传靶区探针组的探针具有少于5kb、至少5kb(例如,至少10kb、20kb或50kb)的足迹。
[0461]
a.高甲基化可变靶区
[0462]
在一些实施方案中,针对表观遗传靶区组的探针包括对一个或更多个高甲基化可变靶区特异性的探针。高甲基化可变靶区可以是上文列出的任何高甲基化可变靶区。例如,在一些实施方案中,对高甲基化可变靶区特异性的探针包括对表1中列出的多于一个基因座(例如,表1中列出的基因座中的至少10%、20%、30%、40%、50%、60%、70%、80%、90%或100%)特异性的探针。在一些实施方案中,对高甲基化可变靶区特异性的探针包括对表2中列出的多于一个基因座(例如,表2中列出的基因座中的至少10%、20%、30%、40%、50%、60%、70%、80%、90%或100%)特异性的探针。在一些实施方案中,对高甲基化可变靶区特异性的探针包括对表1或表2中列出的多于一个基因座(例如,表1或表2中列出的基因座中的至少10%、20%、30%、40%、50%、60%、70%、80%、90%或100%)特异性的探针。在一些实施方案中,对于作为靶区被包括在内的每个基因座,可以有一个或更多个探针,所述探针具有在该基因的转录起始位点和终止密码子(对于选择性剪接的基因为最后的终止密码子)之间结合的杂交位点。在一些实施方案中,一个或更多个探针在所列位置的300bp内(例如,在200bp或100bp内)结合。在一些实施方案中,探针具有与以上列出的位置重叠的杂交位点。在一些实施方案中,对高甲基化靶区特异性的探针包括对高甲基化靶区的一个、两个、三个、四个或五个子集特异性的探针,其集体地显示出在乳腺癌、结肠癌、肾癌、肝癌和肺癌中的一种、两种、三种、四种或五种中的高甲基化。
[0463]
b.低甲基化可变靶区
[0464]
在一些实施方案中,针对表观遗传靶区组的探针包括对一个或更多个低甲基化可变靶区特异性的探针。低甲基化可变靶区可以是上文列出的任何低甲基化可变靶区。例如,对一个或更多个低甲基化可变靶区特异性的探针可以包括针对以下区域的探针:诸如重复元件(例如,line1元件、alu元件、着丝粒串联重复序列、着丝粒周围串联重复序列和卫星dna)和基因间区域,这些区域在健康细胞中通常被甲基化,在肿瘤细胞中可能显示出减少的甲基化。
[0465]
在一些实施方案中,对低甲基化可变靶区特异性的探针包括对重复元件和/或基因间区域特异性的探针。在一些实施方案中,对重复元件特异性的探针包括对line1元件、alu元件、着丝粒串联重复序列、着丝粒周围串联重复序列和/或卫星dna中的一种、两种、三种、四种或五种特异性的探针。
[0466]
对显示出癌症相关低甲基化的基因组区域特异性的示例性探针包括对人类1号染色体的核苷酸8403565-8953708和/或151104701-151106035特异性的探针。在一些实施方案中,对低甲基化可变靶区特异性的探针包括对与人类1号染色体核苷酸8403565-8953708和/或151104701-151106035重叠或包含其的区域特异性的探针。
[0467]
c.ctcf结合区
[0468]
在一些实施方案中,针对表观遗传靶区组的探针包括对ctcf结合区特异性的探针。在一些实施方案中,对ctcf结合区特异性的探针包括对至少10个、20个、50个、100个、200个或500个ctcf结合区、或者10-20个、20-50个、50-100个、100-200个、200-500个或500-1000个ctcf结合区特异性的探针,例如,诸如上文描述的ctcf结合区,或者ctcfbsdb或上文引用的文章cuddapah等人、martin等人或rhee等人中的一项或更多项中的ctcf结合区。在一些实施方案中,针对表观遗传靶区组的探针包含ctcf结合位点的上游和下游区域至少100bp、至少200bp、至少300bp、至少400bp、至少500bp、至少750bp或至少1000bp。
[0469]
d.转录起始位点
[0470]
在一些实施方案中,针对表观遗传靶区组的探针包括对转录起始位点特异性的探针。在一些实施方案中,对转录起始位点特异性的探针包括对至少10个、20个、50个、100个、200个或500个转录起始位点、或者10-20个、20-50个、50-100个、100-200个、200-500个或500-1000个转录起始位点,例如,诸如dbtss中列出的转录起始位点特异性的探针。在一些实施方案中,针对表观遗传靶区组的探针包括针对转录起始位点上游和下游至少100bp、至少200bp、至少300bp、至少400bp、至少500bp、至少750bp或至少1000bp的序列的探针。
[0471]
e.聚焦扩增
[0472]
如以上提到的,尽管聚焦扩增是体细胞突变,但它们可以通过基于读段频率的测序以类似于检测某些表观遗传改变诸如甲基化改变的方法的方式来检测。因此,癌症中可能显示出聚焦扩增的区域可以包括在表观遗传靶区组中,如以上讨论的。在一些实施方案中,对表观遗传靶区组特异性的探针包括对聚焦扩增特异性的探针。在一些实施方案中,对聚焦扩增特异性的探针包括对ar、braf、ccnd1、ccnd2、ccne1、cdk4、cdk6、egfr、erbb2、fgfr1、fgfr2、kit、kras、met、myc、pdgfra、pik3ca和raf1中的一种或更多种特异性的探针。例如,在一些实施方案中,对聚焦扩增特异性的探针包括对前述靶中至少2种、3种、4种、5种、6种、7种、8种、9种、10种、11种、12种、13种、14种、15种、16种、17种或18种中的一种或更多种特异性的探针。
[0473]
f.对照区
[0474]
纳入对照区来帮助数据验证是有用的。在一些实施方案中,对表观遗传靶区组特异性的探针包括对对照甲基化区特异性的探针,所述对照甲基化区预期在基本上所有样品中都是甲基化的。在一些实施方案中,对表观遗传靶区组特异性的探针包括对对照低甲基化区特异性的探针,所述对照低甲基化区预期在基本上所有样品中都是低甲基化的。
[0475]
2.对序列可变靶区特异性的探针
[0476]
针对序列可变靶区组的探针可以包括对癌症中已知经历体细胞突变的多于一个区域特异性的探针。探针可以是对本文描述的任何序列可变靶区组特异性的。在本文(例如,在上文关于捕获组的章节中)详细讨论了示例性序列可变靶区组。
[0477]
在一些实施方案中,序列可变靶区探针组具有至少0.5kb(例如,至少1kb、至少2kb、至少5kb、至少10kb、至少20kb、至少30kb或至少40kb)的足迹。在一些实施方案中,表观遗传靶区探针组具有0.5-100kb(例如,0.5-2kb、2-10kb、10-20kb、20-30kb、30-40kb、40-50kb、50-60kb、60-70kb、70-80kb、80-90kb和90-100kb)范围内的足迹。
[0478]
在一些实施方案中,对序列可变靶区组特异性的探针包括对表3中至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个或70个基因的至少一部分特异性的探针。在一些实施方案中,对序列可变靶区组特异性的探针包括对表3中至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个或70个snv特异性的探针。在一些实施方案中,对序列可变靶区组特异性的探针包括对表3中至少1个、至少2个、至少3个、至少4个、至少5个或6个融合特异性的探针。在一些实施方案中,对序列可变靶区组特异性的探针包括对表3中至少1个、至少2个或3个插入/缺失的至少一部分特异性的探针。在一些实施方案中,对序列可变靶区组特异性的探针包括对表4中至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个、至少70个或73个基因的至少一部分特异性的探针。在一些实施方案中,对序列可变靶区组特异性的探针包括对表4中至少5个、至少10个、至少15个、至少20个、至少25个、至少30个、至少35个、至少40个、至少45个、至少50个、至少55个、至少60个、至少65个、至少70个或73个snv特异性的探针。在一些实施方案中,对序列可变靶区组特异性的探针包括对表4中至少1个、至少2个、至少3个、至少4个、至少5个或6个融合特异性的探针。在一些实施方案中,对序列可变靶区组特异性的探针包括对表4中至少1个、至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、至少14个、至少15个、至少16个、至少17个或18个插入/缺失的至少一部分特异性的探针。在一些实施方案中,对序列可变靶区组特异性的探针包括对表5中至少1个、至少2个、至少3个、至少4个、至少5个、至少6个、至少7个、至少8个、至少9个、至少10个、至少11个、至少12个、至少13个、至少14个、至少15个、至少16个、至少17个、至少18个、至少19个或至少20个基因的至少一部分特异性的探针。
[0479]
在一些实施方案中,对序列可变靶区组特异性的探针包括对来自至少10个、20个、30个或35个癌症相关基因的靶区特异性的探针,所述癌症相关基因诸如akt1、alk、braf、ccnd1、cdk2a、ctnnb1、egfr、erbb2、esr1、fgfr1、fgfr2、fgfr3、foxl2、gata3、gna11、gnaq、gnas、hras、idh1、idh2、kit、kras、med12、met、myc、nfe2l2、nras、pdgfra、pik3ca、ppp2r1a、pten、ret、stk11、tp53和u2af1。
[0480]
e.包含捕获的dna的组合物
[0481]
本文提供了包含捕获的dna的第一群体和第二群体的组合。第一群体可以包含或源自比第二群体具有更大比例的胞嘧啶修饰的dna。第一群体可以包含具有改变的碱基配对特异性的最初存在于dna中的第一核碱基的形式和没有改变的碱基配对特异性的第二核碱基,其中在碱基配对特异性改变之前最初存在于dna中的第一核碱基的形式是修饰或未
修饰的核碱基,第二核碱基是不同于第一核碱基的修饰或未修饰的核碱基,并且在碱基配对特异性改变之前最初存在于dna中的第一核碱基的形式和第二核碱基具有相同的碱基配对特异性。第二群体不包含具有改变的碱基配对特异性的最初存在于dna中的第一核碱基的形式。在一些实施方案中,胞嘧啶修饰是胞嘧啶甲基化。在一些实施方案中,第一核碱基是修饰或未修饰的胞嘧啶,并且第二核碱基是修饰或未修饰的胞嘧啶。第一核碱基和第二核碱基可以是本文在概述中或者关于使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序所讨论的任何核碱基。
[0482]
在一些实施方案中,第一群体包含选自第一组一个或更多个序列标签的序列标签,并且第二群体包含选自第二组一个或更多个序列标签的序列标签,并且第二组序列标签不同于第一组序列标签。序列标签可以包括条形码。
[0483]
在一些实施方案中,第一群体包含受保护的hmc,诸如葡糖基化的hmc。
[0484]
在一些实施方案中,第一群体经历本文讨论的任何转化程序,诸如亚硫酸氢盐转化、ox-bs转化、tab转化、ace转化、tap转化、tapsβ转化或cap转化。在一些实施方案中,第一群体经历对hmc的保护,随后经历mc和/或c的脱氨基。
[0485]
在该组合的一些实施方案中,第一群体包含或源自比第二群体具有更大比例的胞嘧啶修饰的dna,并且第一群体包括第一亚群和第二亚群,并且第一核碱基是修饰或未修饰的核碱基,第二核碱基是不同于第一核碱基的修饰或未修饰的核碱基,并且第一核碱基和第二核碱基具有相同的碱基配对特异性。在一些实施方案中,第二群体不包含第一核碱基。在一些实施方案中,第一核碱基是修饰或未修饰的胞嘧啶,并且第二核碱基是修饰或未修饰的胞嘧啶,任选地其中修饰的胞嘧啶是mc或hmc。在一些实施方案中,第一核碱基是修饰或未修饰的腺嘌呤,并且第二核碱基是修饰或未修饰的腺嘌呤,任选地其中修饰的腺嘌呤是ma。
[0486]
在一些实施方案中,第一核碱基(例如,修饰的胞嘧啶)被生物素化。在一些实施方案中,第一核碱基(例如,修饰的胞嘧啶)是对β-6-叠氮基-葡糖基-5-羟甲基胞嘧啶进行huisgen环加成的产物,该产物包含亲和标记(例如,生物素)。
[0487]
在本文描述的任何组合中,捕获的dna可以包括cfdna。
[0488]
捕获的dna可以具有本文描述的关于捕获组的任何特征,包括例如,对应于序列可变靶区组的dna的浓度比对应于表观遗传靶区组的dna的浓度更大(如以上讨论的针对足迹尺寸进行归一化)。在一些实施方案中,捕获组的dna包含序列标签,所述序列标签可以如本文描述的添加到dna。通常,序列标签的包含导致dna分子不同于它们天然存在的、未加标签的形式。
[0489]
该组合还可以包含本文描述的探针组或测序引物,其中每一个都可不同于天然存在的核酸分子。例如,本文描述的探针组可以包含捕获部分,并且测序引物可以包含非天然存在的标记。
[0490]
f.计算机系统
[0491]
本公开内容的方法可以使用或借助计算机系统来实现。例如,这样的方法可以包括:将样品分区为多于一个子样品,包括第一子样品和第二子样品,其中第一子样品包含比第二子样品具有更大比例的胞嘧啶修饰的dna;使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序,其中第一核碱基是修饰或未修饰的核
碱基,第二核碱基是不同于第一核碱基的修饰或未修饰的核碱基,并且第一核碱基和第二核碱基具有相同的碱基配对特异性;以及以区分第一子样品的dna中的第一核碱基和第二核碱基的方式对第一子样品中的dna和第二子样品中的dna进行测序。
[0492]
图4示出了被编程或以其他方式配置成实现本公开内容的方法的计算机系统401。计算机系统401可以控制样品制备、测序和/或分析的各方面。在一些实例中,计算机系统401被配置为进行样品制备和样品分析,包括核酸测序,例如,根据本文公开的任何方法。
[0493]
计算机系统401包括中央处理单元(cpu,本文中也称为“处理器”和“计算机处理器”)405,其可以是单核或多核处理器或用于并行处理的多于一个处理器。计算机系统401还包括存储器或存储器位置410(例如,随机存取存储器、只读存储器、闪速存储器)、电子存储单元415(例如,硬盘)、用于与一个或更多个其他系统进行通信的通信接口420(例如,网络适配器)和外围设备425,诸如高速缓冲存储器(cache)、其他存储器、数据存储和/或电子显示适配器。存储器410、储存单元415、接口420和外围设备425与cpu 405通过通信网络或总线(实线路)诸如主板(motherboard)通信。存储单元415可以是用于存储数据的数据存储单元(或数据储存库)。计算机系统401可以借助于通信接口420可操作地耦合至计算机网络430。计算机网络430可以是因特网(internet)、内联网和/或外联网、或与因特网通信的内联网和/或外联网。在一些情况下,计算机网络430为电信和/或数据网络。计算机网络430可以包括一个或更多个计算机服务器,这可以启动分布式计算,诸如云计算。在一些情况下,借助于计算机系统401,计算机网络430可以实现对等网络(peer-to-peer network),其可以启动耦合至计算机系统401的设备作为客户端或服务器运行。
[0494]
cpu 405可以执行一系列的机器可读指令,该机器可读指令可以以程序或软件来体现。指令可以被存储于存储器位置,诸如存储器410中。由cpu 405进行的操作的实例可以包括读取、解码、执行和写回。
[0495]
存储单元415可以存储文件,诸如驱动程序、库和保存的程序。存储单元415可以存储用户生成的程序和记录的会话以及与程序相关的输出。存储单元415可以存储用户数据,例如,用户偏好和用户程序。在一些情况下,计算机系统401可以包括一个或更多个另外的数据存储单元,该另外的数据存储单元在计算机系统401的外部,诸如位于通过内联网或因特网与计算机系统401通信的远程服务器上。可以使用例如通信网络或物理数据传输器(例如,使用硬盘驱动器、拇指驱动器或其他数据存储机制)将数据从一个位置传输到另一个位置。
[0496]
计算机系统401可以与一个或更多个远程计算机系统通过网络430进行通信。对于实施方案,计算机系统401可以与用户(例如,操作者)的远程计算机系统进行通信。远程计算机系统的实例包括个人计算机(例如,便携式pc)、板式(slate)或平板pc(例如,ipad、galaxy tab)、电话、智能电话(例如,iphone、android支持的设备、)或个人数字助手。用户可以通过网络430访问计算机系统401。
[0497]
如本文描述的方法可以通过机器(例如,计算机处理器)可执行代码的方式实现,该机器可进行代码被存储在计算机系统401的电子存储位置,诸如,例如存储器410或电子存储单元415上。机器可执行代码或机器可读代码可以以软件的形式提供。在使用期间,代码可以由处理器405执行。在一些情况下,代码可以从存储单元415检索并存储在存储器410上,以便于处理器405即时访问。在一些情况下,可以排除电子存储单元415,而将机器可执
行指令存储于存储器410上。
[0498]
在一方面,本公开内容提供了包含计算机可执行指令的非暂时性计算机可读介质,当该计算机可执行指令由至少一个电子处理器执行时,进行包括以下的方法的至少一部分:从测试受试者收集cfdna;从cfdna捕获多于一个靶区组,其中该多于一个靶区组包括序列可变靶区组和表观遗传靶区组,由此产生cfdna分子的捕获组;对捕获的cfdna分子进行测序,其中将序列可变靶区组的捕获的cfdna分子测序到比表观遗传靶区组的捕获的cfdna分子更大的测序深度;获得由核酸测序仪从对捕获的cfdna分子进行测序生成的多于一个序列读段;将多于一个序列读段映射到一个或更多个参考序列以生成映射的序列读段;以及处理对应于序列可变靶区组和表观遗传靶区组的映射的序列读段,以确定受试者患有癌症的可能性。
[0499]
代码可以被预编译并配置成用于与具有适于执行该代码的处理器的机器一起使用或可以在运行时间期间被编译。代码可以以编程语言的形式提供,该编程语言可以被选择使得代码能够以预编译的或按原来编译(as-compiled)的方式被执行。
[0500]
本文提供的系统和方法的方面,诸如计算机系统401,可以以编程来体现。技术的多个方面可以被认为是“产品”或“制品”,所述产品”或“制品”通常呈在某种类型的机器可读介质上携带或在所述机器可读介质中体现的机器(或处理器)可执行代码和/或相关的数据的形式。机器可执行代码可以被存储于电子存储单元诸如存储器(例如,只读存储器、随机存取存储器、闪速存储器)或硬盘上。“存储”型介质可以包括计算机、处理器等任何或所有有形存储器,或其相关的模块,诸如多种半导体存储器、磁带驱动器、磁盘驱动器等,其可以在任何时间为软件编程提供非暂时性存储。
[0501]
软件的所有或部分有时可以通过互联网或多种其他电信网络通信。例如,这样的通信可以使软件能够从一个计算机或处理器加载到另一个计算机或处理器,例如,从管理服务器或主机加载到应用服务器的计算机平台。因此,可以携带软件元件的另一类型的介质包括诸如那些在本地设备之间跨物理界面、通过有线和光纤陆线网络以及在多种空中链路(air-link)上使用的光波、电波和电磁波。携带此类波的物理元件,诸如有线或无线链路、光链路等,也可被认为是携带软件的介质。如本文使用的,除非被限制为非暂时性的、有形的“存储”介质,否则术语诸如计算机或机器“可读介质”是指参与将指令提供至处理器以便执行的任何介质。
[0502]
因此,机器可读介质,诸如计算机可执行代码,可以采取许多形式,包括但不限于有形存储介质、载波介质或物理传输介质。非易失性存储介质包括,例如光盘或磁盘,诸如任何一个或多于一个计算机等中的任何存储装置,诸如如附图中显示出的可以用于实现数据库等的存储装置。易失性存储介质包括动态存储器,诸如这样的计算机平台的主存储器。有形的传输介质包括同轴电缆;铜线和光纤,包括构成计算机系统内的总线的导线。载波传输介质可以采取电信号或电磁信号或者声波或光波的形式,诸如在射频(rf)和红外(ir)数据通信期间生成的那些。因此,计算机可读介质的常见形式包括例如:软盘(floppy disk)、软磁盘(flexible disk)、硬盘、磁带、任何其他磁介质、cd-rom、dvd或dvd-rom、任何其他光学介质、穿孔卡片、纸带、具有孔模式的任何其他物理存储介质、ram、rom、prom和eprom、flash-eprom、任何其他存储器芯片或盒、传输数据或指令的载波、传输此类载波的缆线或链路,或者计算机可以从其读取编程代码和/或数据的任何其他介质。计算机可读介质的这
些形式中的许多形式可以参与向处理器传送一个或更多个指令的一个或更多个序列以用于执行。
[0503]
计算机系统401可以包括电子显示器或与之通信,该电子显示器包括用户界面(ui),以便提供例如样品分析的一个或更多个结果。ui的实例包括但不限于图形用户界面(gui)和基于网络的用户界面。
[0504]
涉及计算机系统和网络、数据库和计算机程序产品的另外的细节也在以下中提供:例如,peterson,computer networks:a systems approach,morgan kaufmann,第5版(2011);kurose,computer networking:a top-down approach,pearson,第7版(2016);elmasri,fundamentals of database systems,addison wesley,第6版(2010);coronel,database systems:design,implementation,&management,cengage learning,第11版(2014);tucker,programming languages,mcgraw-hill science/engineering/math,第2版(2006);和rhoton,cloud computing architected:solution design handbook,recursive press(2011),其中每一项通过引用以其整体并入本文。
[0505]
g.应用
[0506]
1.癌症和其他疾病
[0507]
本方法可以用于诊断受试者中状况特别是癌症的存在,以表征状况(例如,对癌症进行分期或确定癌症的异质性),监测状况对治疗的响应,实现对状况发展或状况后续进程的风险的预后。本公开内容也可以用于确定特定治疗选择的效力。如果治疗是成功的,则成功的治疗选择可能随着更多的癌症可能死亡并且使dna脱落而增加受试者的血液中检测到的拷贝数变异或罕见突变的量。在其他实例中,这可能不会发生。在另一实例中,也许某些治疗选择可能与癌症随时间推移的遗传谱相关。这种相关性可以用于选择疗法。
[0508]
另外,如果观察到癌症在治疗之后处于缓解中,本方法可以用于监测残留疾病或疾病的复发。
[0509]
在一些实施方案中,本文公开的方法和系统可以基于将核酸变异分类为体细胞来源或种系来源而用于鉴定定制或靶向的疗法以治疗患者的特定疾病或状况。通常,所考虑的疾病是一种类型的癌症。这样的癌症的非限制性实例包括胆道癌、膀胱癌、移行细胞癌、尿路上皮癌、脑癌、神经胶质瘤、星形细胞瘤、乳腺癌、化生癌、宫颈癌、宫颈鳞状细胞癌、直肠癌、结肠直肠癌、结肠癌、遗传性非息肉性结肠直肠癌、结肠腺癌、胃肠间质瘤(gist)、子宫内膜癌、子宫内膜间质肉瘤、食管癌、食管鳞状细胞癌、食管腺癌、眼黑素瘤、葡萄膜黑素瘤、胆囊癌、胆囊腺癌、肾细胞癌、透明细胞肾细胞癌(clear cell renal cell carcinoma)、移行细胞癌、尿路上皮癌、肾母细胞瘤、白血病、急性淋巴细胞白血病(all)、急性髓性白血病(aml)、慢性淋巴细胞白血病(cll)、慢性髓性白血病(cml)、慢性粒单核细胞白血病(cmml)、肝癌(liver cancer)、肝癌(liver carcinoma)、肝细胞瘤、肝细胞癌、胆管癌、肝母细胞瘤、肺癌、非小细胞肺癌(nsclc)、间皮瘤、b细胞淋巴瘤、非霍奇金淋巴瘤、弥漫性大b细胞淋巴瘤、套细胞淋巴瘤、t细胞淋巴瘤、非霍奇金淋巴瘤、前体t淋巴母细胞淋巴瘤/白血病、外周t细胞淋巴瘤、多发骨髓瘤、鼻咽癌(npc)、神经母细胞瘤、口咽癌、口腔鳞状细胞癌、骨肉瘤、卵巢癌、胰腺癌、胰腺导管腺癌、假乳头状肿瘤、泡细胞癌。前列腺癌、前列腺腺癌、皮肤癌、黑素瘤、恶性黑素瘤、皮肤黑素瘤、小肠癌、胃癌(stomach cancer)、胃癌(gastric carcinoma)、胃肠间质瘤(gist)、子宫癌或子宫肉瘤。癌症的类型和/或分期可以
根据遗传变异检测,包括突变、罕见突变、插入/缺失、拷贝数变异、颠换、易位、倒位、缺失、非整倍性、部分非整倍性、多倍性、染色体不稳定性、染色体结构改变、基因融合、染色体融合、基因截短、基因扩增、基因复制、染色体损伤、dna损伤、核酸化学修饰的异常改变、表观遗传模式的异常改变和核酸5-甲基胞嘧啶的异常改变。
[0510]
遗传数据还可以用于表征特定形式的癌症。癌症在组成和分期两方面通常是异质性的。遗传谱数据可以允许表征癌症的具体亚型,该表征在该具体亚型的诊断或治疗中可能是重要的。该信息还可以为受试者或从业者提供关于具体类型癌症的预后的线索,并且允许受试者或从业者根据疾病的进展调整治疗选择。一些癌症可以进展而变得更具侵袭性和遗传不稳定性。其他癌症可以保持良性的、非活动的、或休眠的。本公开内容的系统和方法可以用于确定疾病进展。
[0511]
此外,本公开内容的方法可以用于表征受试者的异常状况的异质性。这样的方法可以包括,例如生成源自受试者的细胞外多核苷酸的遗传谱,其中所述遗传谱包括由拷贝数变异和罕见突变分析得到的多于一个数据。在一些实施方案中,异常状况是癌症。在一些实施方案中,异常状况可以是导致异质性基因组群体的状况。在癌症的实例中,已知一些肿瘤包含处于癌症的不同分期的肿瘤细胞。在其他实例中,异质性可以包括疾病的多个病灶。再次,在癌症的实例中,可以存在多个肿瘤病灶,或许其中一个或更多个病灶为已从原发部位扩散的转移的结果。
[0512]
本方法可以用于产生或剖析为来源于异质性疾病中不同细胞遗传信息的总和的指纹图谱或数据集。该数据集可以包括单独的或组合的拷贝数变异、表观遗传变异和突变分析。
[0513]
本方法可以用于诊断、预后、监测或观察癌症或其他疾病。在一些实施方案中,本文的方法不涉及胎儿的诊断、预后或监测胎儿,并因此不涉及非侵入性产前测试。在其他实施方案中,这些方法可以用于妊娠受试者中以诊断、预后、监测或观察未出生受试者中的癌症或其他疾病,所述未出生受试者的dna和其他多核苷酸可以与母体分子共循环。
[0514]
任选地使用本文公开的方法和系统评估的其它基于遗传的疾病、紊乱或状况的非限制性实例包括软骨发育不全、α-1抗胰蛋白酶缺乏症、抗磷脂综合征、孤独症、常染色体显性多囊肾病、夏科-马里-图思病(cmt)、猫叫综合征、克罗恩病、囊性纤维化、dercum病、唐氏综合征、duane综合征、杜兴氏肌营养不良症、因子v leiden易栓症、家族性高胆固醇血症、家族性地中海热、脆性x综合征、戈谢病、血色素沉着病、血友病、全前脑畸形、亨廷顿病、克兰费尔特综合征、马方综合征、强直性肌营养不良、神经纤维瘤病、努南综合征、成骨不全、帕金森病、苯丙酮尿症、poland异常、卟啉症、早老症、视网膜色素变性、重症联合免疫缺陷病(scid)、镰状细胞病、脊髓性肌萎缩症、泰-萨克斯病、地中海贫血、三甲基胺尿症、特纳综合征、颚心脸综合征(velocardiofacial syndrome)、wagr综合征、威尔逊病等。
[0515]
在一些实施方案中,本文描述的方法包括在先前被诊断为患有癌症的受试者的先前癌症治疗之后的预选时间点,使用如本文描述获得的序列信息组检测来源于或源自肿瘤细胞的dna的存在或不存在。该方法还可以包括确定癌症复发评分,所述癌症复发评分指示来源于或源自测试受试者的肿瘤细胞的dna的存在或不存在。
[0516]
在确定了癌症复发评分的情况下,可以进一步使用它来确定癌症复发状态。例如,当癌症复发评分高于预定阈值时,癌症复发状态可能处于癌症复发风险。例如,当癌症复发
评分低于预定阈值时,癌症复发状态可能处于低或较低的癌症复发风险。在特定实施方案中,等于预定阈值的癌症复发评分可以得到处于癌症复发风险或者处于低或较低的癌症复发风险的癌症复发状态。
[0517]
在一些实施方案中,将癌症复发评分与预定的癌症复发阈值进行比较,并且当癌症复发评分高于癌症复发阈值时,将测试受试者分类为后续癌症治疗的候选者,或者当癌症复发评分低于癌症复发阈值时,将测试受试者分类为治疗的非候选者。在特定实施方案中,等于癌症复发阈值的癌症复发评分可以导致分类为后续癌症治疗的候选者或治疗的非候选者。
[0518]
以上讨论的方法还可以包括任何相容特征或在本文其他处(包括关于确定测试受试者的癌症复发风险和/或将测试受试者分类为后续癌症治疗的候选者的方法的章节中)阐述的特征。
[0519]
2.确定测试受试者的癌症复发风险和/或将测试受试者分类为后续癌症治疗的候选者的方法
[0520]
在一些实施方案中,本文提供的方法是确定测试受试者的癌症复发风险的方法。在一些实施方案中,本文提供的方法是将测试受试者分类为后续癌症治疗的候选者的方法。
[0521]
这样的方法中的任一种可以包括在对测试受试者进行一个或更多个先前的癌症治疗之后的一个或更多个预选时间点从被诊断为患有癌症的测试受试者收集dna(例如,来源于或源自肿瘤细胞)。受试者可以是本文描述的任何受试者。dna可以是cfdna。dna可以从组织样品中获得。
[0522]
这样的方法中的任一种可以包括从来自受试者的dna中捕获多于一个靶区组,其中多于一个靶区组包括序列可变靶区组和表观遗传靶区组,由此产生dna分子的捕获组。捕获步骤可以根据本文其他处描述的任何实施方案来进行。
[0523]
在这样的方法中的任一种中,先前的癌症治疗可包括手术、施用治疗组合物和/或化学疗法。
[0524]
这样的方法中的任一种可以包括对捕获的dna分子进行测序,由此产生序列信息组。可以将序列可变靶区组的捕获的dna分子测序到比表观遗传靶区组的捕获的dna分子更大的测序深度。
[0525]
这样的方法中的任一种可以包括在预选的时间点使用序列信息组检测来源于或源自肿瘤细胞的dna的存在或不存在。对来源于或源自肿瘤细胞的dna的存在或不存在的检测可以根据本文其他处描述的它的任何实施方案来进行。
[0526]
确定测试受试者的癌症复发风险的方法可以包括确定癌症复发评分,所述癌症复发评分指示来源于或源自测试受试者的肿瘤细胞的dna的存在或不存在或者量。癌症复发评分可以进一步用于确定癌症复发状态。例如,当癌症复发评分高于预定阈值时,癌症复发状态可能处于癌症复发风险。例如,当癌症复发评分低于预定阈值时,癌症复发状态可能处于低或较低的癌症复发风险。在特定实施方案中,等于预定阈值的癌症复发评分可以得到处于癌症复发风险或者处于低或较低的癌症复发风险的癌症复发状态。
[0527]
将测试受试者分类为后续癌症治疗的候选者的方法可以包括将测试受试者的癌症复发评分与预定癌症复发阈值进行比较,从而当癌症复发评分高于癌症复发阈值时将测
试受试者分类为后续癌症治疗的候选者,或者当癌症复发评分低于癌症复发阈值时将测试受试者分类为治疗的非候选者。在特定实施方案中,等于癌症复发阈值的癌症复发评分可以得到作为后续癌症治疗的候选者或治疗的非候选者的分类。在一些实施方案中,后续癌症治疗包括化学疗法或施用治疗组合物。
[0528]
这样的方法中的任一种可以包括基于癌症复发评分确定测试受试者的无疾病存活(dfs)期;例如,dfs期可以是1年、2年、3年、4年、5年或10年。
[0529]
在一些实施方案中,序列信息组包括序列可变靶区序列,并且确定癌症复发评分可以包括确定至少第一子评分,所述第一子评分指示序列可变靶区序列中存在的snv、插入/缺失、cnv和/或融合的量。
[0530]
在一些实施方案中,序列可变靶区中选自1个、2个、3个、4个或5个的突变数目足以使第一子评分导致癌症复发评分被分类为癌症复发阳性。在一些实施方案中,突变数目选自1个、2个或3个。
[0531]
在一些实施方案中,序列信息组包括表观遗传靶区序列,并且确定癌症复发评分包括确定指示分子(从表观遗传靶区序列中获得)的量的第二子评分,所述分子代表不同于在来自健康受试者的相应样品中发现的dna(例如,在来自健康受试者的血液样品中发现的cfdna,或在来自健康受试者的组织样品中发现的dna,其中组织样品是与从测试受试者获得的组织相同类型的组织)的表观遗传状态。这些异常分子(即,具有不同于来自健康受试者的相应样品中发现的dna的表观遗传状态的分子)可以和与癌症相关的表观遗传改变一致,例如,高甲基化可变靶区的甲基化和/或片段化可变靶区的被扰动的片段化,其中“被扰动”意指不同于来自健康受试者的相应样品中发现的dna。
[0532]
在一些实施方案中,大于或等于0.001%-10%范围内的值的指示高甲基化可变靶区组中的高甲基化和/或片段化可变靶区组中的异常片段化的对应于高甲基化可变靶区组和/或片段化可变靶区组的分子比例足以使第二子评分被分类为癌症复发阳性。范围可为0.001%-1%、0.005%-1%、0.01%-5%、0.01%-2%或0.01%-1%。
[0533]
在一些实施方案中,这样的方法中的任一种可以包括根据指示一个或更多个指示来自肿瘤细胞的来源的特征的序列信息组中的分子分数确定肿瘤dna分数。这可以用于对应于表观遗传靶区中的一些或所有的分子,例如,包括高甲基化可变靶区和片段化可变靶区(高甲基化可变靶区的高甲基化和/或片段化可变靶区的异常片段化可以被认为指示来自肿瘤细胞的来源)中的一个或两者。这可以用于对应于序列可变靶区的分子,例如,包含与癌症一致的改变(诸如snv、插入/缺失、cnv和/或融合)的分子。肿瘤dna分数可以基于对应于表观遗传靶区的分子和对应于序列可变靶区的分子的组合来确定。
[0534]
癌症复发评分的确定可以至少部分地基于肿瘤dna分数,其中大于10-11
至1或10-10
至1的范围内的阈值的肿瘤dna分数足以使癌症复发评分被分类为癌症复发阳性。在一些实施方案中,大于或等于10-10
至10-9
、10-9
至10-8
、10-8
至10-7
、10-7
至10-6
、10-6
至10-5
、10-5
至10-4
、10-4
至10-3
、10-3
至10-2
或10-2
至10-1
的范围内的阈值的肿瘤dna分数足以使癌症复发评分被分类为癌症复发阳性。在一些实施方案中,大于至少10-7
的阈值的肿瘤dna分数足以使癌症复发评分被分类为癌症复发阳性。可以基于累积概率来确定肿瘤dna分数大于阈值,诸如对应于任何前述实施方案的阈值。例如,如果肿瘤分数大于任何前述范围中的阈值的累积概率超过至少0.5、0.75、0.9、0.95、0.98、0.99、0.995或0.999的概率阈值,则认为样品为阳
性。在一些实施方案中,概率阈值为至少0.95,诸如0.99。
[0535]
在一些实施方案中,序列信息组包括序列可变靶区序列和表观遗传靶区序列,并且确定癌症复发评分包括确定指示序列可变靶区序列中存在的snv、插入/缺失、cnv和/或融合的量的第一子评分和指示表观遗传靶区序列中异常分子的量的第二子评分,以及组合第一子评分和第二子评分以提供癌症复发评分。在组合第一子评分和第二子评分的情况下,它们可以通过以下方式来组合:将阈值独立地应用于每个子评分(例如,在序列可变靶区中大于预定的突变数目(例如》1),并且在表观遗传靶区中大于预定的异常分子(即,具有不同于在来自健康受试者的相应样品中发现的dna的表观遗传状态的分子;例如肿瘤)分数),或者训练机器学习分类器以基于多于一个阳性训练样品和阴性训练样品来确定状态。
[0536]
在一些实施方案中,在-4至2或-3至1的范围内的组合评分的值足以使癌症复发评分被分类为癌症复发阳性。
[0537]
在其中癌症复发评分被分类为癌症复发阳性的任何实施方案中,受试者的癌症复发状态可能处于癌症复发的风险和/或可以将受试者分类为后续癌症治疗的候选者。
[0538]
在一些实施方案中,癌症是本文其他处描述的癌症类型中的任一种,例如,结肠直肠癌。
[0539]
3.治疗和相关管理
[0540]
在某些实施方案中,本文公开的方法涉及鉴于核酸变异为体细胞来源或种系来源的状态,鉴定定制疗法并向患者施用定制疗法。在一些实施方案中,基本上任何癌症疗法(例如,手术疗法、放射疗法、化学疗法和/或类似疗法)都可以被包括为这些方法的一部分。通常,定制疗法包括至少一种免疫疗法(或免疫治疗剂)。免疫疗法通常是指增强针对特定癌症类型的免疫应答的方法。在某些实施方案中,免疫疗法是指增强针对肿瘤或癌症的t细胞应答的方法。
[0541]
在某些实施方案中,来自受试者的样品的核酸变异为体细胞来源或种系来源的状态可以与来自参考群体的比较器结果(comparator results)的数据库进行比较,以鉴定用于该受试者的定制或靶向疗法。通常,参考群体包括与测试受试者具有相同癌症或疾病类型的患者和/或正在接受或已经接受与测试受试者相同治疗的患者。当核酸变异和比较器结果满足某些分类标准(例如,基本或近似匹配)时,可以鉴定定制或靶向治疗(或多于一种治疗)。
[0542]
在某些实施方案中,本文描述的定制疗法通常为胃肠外(例如,静脉内或皮下)施用。含有免疫治疗剂的药物组合物通常静脉内施用。某些治疗剂是口服施用的。然而,定制疗法(例如,免疫治疗剂等)也可以通过以下方法施用,诸如例如,含服、舌下、直肠、阴道、尿道内、表面(topical)、眼内、鼻内和/或耳内,所述施用可以包括片剂、胶囊、颗粒、水性悬浮液、凝胶、喷雾剂、栓剂、油膏(salve)、软膏(ointment)等。
[0543]
虽然本文已经示出和描述了本发明的优选实施方案,但对于本领域技术人员将明显的是,此类实施方案仅通过示例的方式提供。并不意图本发明限于本说明书中提供的特定实例。虽然已参考以上提及的说明书描述了本发明,但本文实施方案的描述和说明并不意图以限制性的意义来解释。在不偏离本发明的情况下,本领域技术人员现在将想到许多变化、改变和替换。此外,应当理解,本发明的所有方面并不限于本文根据各种条件和变量阐述的具体描述、配置或相对比例。应当理解,在实践本发明时可以采用本文描述的本公开
内容的实施方案的各种替代选择。因此设想本公开内容还应涵盖任何此类替代选择、修改、变化或等同物。所附权利要求意图界定本发明的范围,并且从而涵盖在这些权利要求范围内的方法和结构及其等同物。
[0544]
虽然为了清楚与理解的目的,已经通过图示和实例的方式对前述公开内容进行了一些详细描述,但是本领域普通技术人员通过阅读本公开内容将会清楚,在不偏离本公开内容的真实范围的情况下,可以进行形式和细节上的多种改变,并且可以在所附权利要求书的范围内实施。例如,所有方法、系统、计算机可读介质和/或组件特征、步骤、元件或其他方面都可以以多种组合来使用。
[0545]
h.试剂盒
[0546]
还提供了包含如本文描述的组合物的试剂盒。试剂盒可用于进行如本文描述的方法。在一些实施方案中,试剂盒包含用于将样品分区为如本文描述的多于一个子样品的第一试剂,诸如本文其他处描述的任何分区试剂。在一些实施方案中,试剂盒包含第二试剂(例如,用于将核碱基诸如胞嘧啶或甲基化胞嘧啶转化为不同核碱基的本文其他处描述的任何试剂),所述第二试剂用于使第一子样品经历不同地影响第一子样品的dna中的第一核碱基和dna中的第二核碱基的程序,其中第一核碱基是修饰或未修饰的核碱基,第二核碱基是不同于第一核碱基的修饰或未修饰的核碱基,并且第一核碱基和第二核碱基具有相同的碱基配对特异性。试剂盒可以包含第一试剂和第二试剂以及下文和/或本文其他处讨论的另外的要素。
[0547]
试剂盒还可以包含多于一个寡核苷酸探针,所述寡核苷酸探针选择性地与至少5个、6个、7个、8个、9个、10个、20个、30个、40个或所有选自由以下组成的组的基因杂交:alk、apc、braf、cdkn2a、egfr、erbb2、fbxw7、kras、myc、notch1、nras、pik3ca、pten、rbi、tp53、met、ar、abl1、akt1、atm、cdh1、csfir、ctnnbl、erbb4、ezh2、fgfrl、fgfr2、fgfr3、flt3、gna11、gnaq、gnas、hnf1a、hras、idh1、idh2、jak2、jak3、kdr、kit、mlh1、mpl、npm1、pdgfra、proc、ptpn11、ret,smad4、smarcb1、smo、src、stk11、vhl、tert、ccnd1、cdk4、cdkn2b、raf1、brca1、ccnd2、cdk6、nf1、tp53、arid 1a、brca2、ccne1、esr1、rit1、gata3、map2k1、rheb、ros1、araf、map2k2、nfe2l2、rhoa和ntrkl。寡核苷酸探针可以选择性杂交的基因的数目可以不同。例如,基因的数目可以包括1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53或54。试剂盒可以包含容器,所述容器包含用于进行本文描述的任何方法的多于一种寡核苷酸探针和说明书。
[0548]
寡核苷酸探针可以选择性地与基因(例如,至少5个基因)的外显子区杂交。在一些情况下,寡核苷酸探针可以选择性地与基因(例如,至少5个基因)的至少30个外显子杂交。在一些情况下,多于一种探针可以选择性地与至少30个外显子中的每一个杂交。与每个外显子杂交的探针可以具有与至少1种其他探针重叠的序列。在一些实施方案中,寡探针可以选择性地与本文公开的基因的非编码区(例如,基因的内含子区)杂交。寡探针还可以选择性地与包含本文公开的基因的外显子区和内含子区两者的基因的区域杂交。
[0549]
寡核苷酸探针可以靶向任何数目的外显子。例如,可以靶向至少1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25个、30个、35个、40个、45个、50个、55个、60个、65个、70个、75
个、80个、85个、90个、95个、100个、105个、110个、115个、120个、125个、130个、135个、140个、145个、150个、155个、160个、165个、170个、175个、180个、185个、190个、195个、200个、205个、210个、215个、220个、225个、230个、235个、240个、245个、250个、255个、260个、265个、270个、275个、280个、285个、290个、个、295个、300个、400个、500个、600个、700个、800个、900个、1,000个或更多个外显子。
[0550]
试剂盒可以包含至少4种、5种、6种、7种或8种具有不同分子条形码和相同样品条形码的不同文库衔接子。文库衔接子可以不是测序衔接子。例如,文库衔接子不包含流动池序列或允许形成用于测序的发夹环的序列。分子条形码和样品条形码的不同变化形式和组合在全文中描述,并适用于试剂盒。此外,在一些情况下,衔接子不是测序衔接子。另外,与试剂盒一起提供的衔接子还可以包括测序衔接子。测序衔接子可以包含与一种或更多种测序引物杂交的序列。测序衔接子还可以包含与固体支持物杂交的序列,例如流动池序列。例如,测序衔接子可以是流动池衔接子。测序衔接子可以附接到多核苷酸片段的一端或两端。在一些情况下,试剂盒可以包含至少8种具有不同分子条形码和相同样品条形码的不同文库衔接子。文库衔接子可以不是测序衔接子。试剂盒还可以包含测序衔接子,所述测序衔接子具有选择性地与文库衔接子杂交的第一序列和选择性地与流动池序列杂交的第二序列。在另一实例中,测序衔接子可以是发夹形的。例如,发夹形衔接子可以包含互补的双链部分和环部分,其中双链部分可以附接(例如,连接)到双链多核苷酸。发夹形测序衔接子可以附接到多核苷酸片段的两端以产生环状分子,其可被多次测序。测序衔接子可以从端到端包含上至10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25个、26个、27个、28个、29个、30个、31个、32个、33个、34个、35个、36个、37个、38个、39个、40个、41个、42个、43个、44个、45个、46个、47个、48个、49个、50个、51个、52个、53个、54个、55个、56个、57个、58个、59个、60个、61个、62个、63个、64个、65个、66个、67个、68个、69个、70个、71个、72个、73个、74个、75个、76个、77个、78个、79个、80个、81个、82个、83个、84个、85个、86个、87个、88个、89个、90个、91个、92个、93个、94个、95个、96个、97个、98个、99个、100个或更多个碱基。测序衔接子可以从端到端包含20-30个、20-40个、30-50个、30-60个、40-60个、40-70个、50-60个、50-70个碱基。在特定实例中,测序衔接子可以从端到端包含20-30个碱基。在另一实例中,测序衔接子可以从端到端包含50-60个碱基。测序衔接子可以包含一种或更多种条形码。例如,测序衔接子可以包含样品条形码。样品条形码可以包含预定序列。样品条形码可用于鉴定多核苷酸的来源。样品条形码可以是至少1个、2个、3个、4个、5个、6个、7个、8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25个或更多个(或如全文中描述的任何长度)核酸碱基,例如至少8个碱基。条形码可以是连续或非连续的序列,如上文描述的。
[0551]
文库衔接子可以是平末端和y形的,并且长度可以小于或等于40个核酸碱基。文库衔接子的其他变化形式可以在全文中找到,并适用于该试剂盒。
[0552]
本文引用的所有专利、专利申请、网站、其他出版物或文件、登录号等都为了所有目的通过引用以其整体并入,其程度如同每个单独的项目都被具体且单独地指示通过引用如此并入一样。如果序列的不同版本在不同时间与一个登记号关联,则意指在本技术的有效申请日时与该登记号关联的版本。如果适用的话,有效提交日期意指真实提交日期或提及该登记号的优先权申请的提交日期中较早的一个。同样,如果出版物、网站等的不同版本
在不同时间发布,则意指在本技术的实际提交日期最近发布的版本,除非另有指示。
[0553]
iii.实施例
[0554]
实施例1:分析cfdna以检测肿瘤的存在/不存在
[0555]
在夸登特健康公司(guardant health)(redwood city,ca,usa)通过基于血液的ngs测定分析一组患者样品以检测癌症的存在/不存在。从这些患者的血浆中提取cfdna。然后将患者样品的cfdna与甲基结合结构域(mbd)缓冲液和与mbd蛋白缀合的磁珠组合,并孵育过夜。在该孵育期间,甲基化cfdna(如果在cfdna样品中存在的话)与mbd蛋白结合。用含有递增盐浓度的缓冲液将非甲基化或甲基化较少的dna从珠上洗下。最后,使用高盐缓冲液将重度甲基化的dna从mbd蛋白洗下。这些洗涤产生甲基化递增的cfdna的三种分区(低甲基化分区、残留甲基化分区和高甲基化分区)。在本实施例中,高甲基化分区中的cfdna分子经历称为hmc-seal/5hmc-seal的程序(han,d.a highly sensitive and robust method for genome-wide 5hmc profiling of rare cell populations.mol cell.2016;63(4):711-719),该程序标记hmc残基(在高甲基化分区的cfdna分子中)以形成β-6-叠氮基-葡糖基-5-羟甲基胞嘧啶,并且然后通过huisgen环加成附接生物素部分,随后使用生物素结合剂将生物素化dna与其他dna分离,从而将高甲基化分区中具有hmc残基的cfdna分子分离到第四分区(hmc分区)中。然后,清洗四个分区中的cfdna分子以去除盐,并浓缩以准备用于文库制备的酶促步骤。
[0556]
在将分区中的cfdna浓缩后,将分区的cfdna的末端突出端延伸,并在延伸期间通过聚合酶将腺苷残基添加到cfdna片段的3’末端。将每个片段的5’末端磷酸化。这些修饰使分区的cfdna可连接。添加dna连接酶和衔接子以将每个分区的cfdna分子的每个末端与衔接子连接。这些衔接子含有非独特分子条形码,并且每个分区与具有非独特分子条形码的衔接子连接,所述非独特分子条形码与用于其他分区的衔接子中的条形码是可区分的。连接后,将四个分区汇集在一起并通过pcr进行扩增。
[0557]
在pcr之后,洗涤扩增的dna并在富集之前浓缩。在浓缩后,将扩增的dna与盐缓冲液和生物素化rna探针(包括针对序列可变靶区组的探针和针对表观遗传靶区组的探针)组合,并将该混合物孵育过夜。针对序列可变区组的探针具有约50kb的足迹,并且针对表观遗传靶区组的探针具有约500kb的足迹。针对序列可变靶区组的探针包括靶向表3-5中鉴定的至少一个基因子集的寡核苷酸,并且针对表观遗传靶区组的探针包括靶向以下选择的寡核苷酸:高甲基化可变靶区、低甲基化可变靶区、ctcf结合靶区、转录起始位点靶区、聚焦扩增靶区和甲基化对照区。
[0558]
生物素化rna探针(与dna杂交)被链霉亲和素磁珠捕获,并通过一系列基于盐的洗涤与未捕获的扩增的dna分离,从而富集样品。富集后,使用illumina novaseq测序仪对富集样品的等分试样进行测序。然后使用生物信息学工具/算法分析由测序仪生成的序列读段。分子条形码用于鉴定独特分子以及将样品解卷积为差异性mbd分区的分子。本实施例中描述的方法,除了基于分子的分区提供关于分子的总体甲基化水平(即,甲基化胞嘧啶残基)的信息之外,还可以基于高甲基化分区的进一步分区以产生hmc分区来提供关于甲基化胞嘧啶类型(即mc或hmc)的身份和/或位置的更高分辨率信息。序列可变靶区序列通过检测可以判定的基因组改变(诸如snv、插入、缺失和融合)来分析,所述判定具有区分真实肿瘤变异与技术错误(例如,pcr错误、测序错误)的足够的支持。独立地分析表观遗传靶区序列
以检测与正常细胞相比在癌症中显示出差异性甲基化的区域中甲基化的cfdna分子。最后,将两个分析的结果结合以产生最终的肿瘤存在/不存的判定。
[0559]
实施例2:以单核苷酸分辨率分析来自健康受试者和患有早期结肠直肠癌的受试者的cfdna样品中的甲基化
[0560]
对来自健康受试者和患有早期结肠直肠癌的受试者的cfdna样品进行如下分析。使用mbd将cfdna分区以提供高甲基化分区、中等甲基化分区和低甲基化分区。将每个分区中分区的dna连接到衔接子(其中衔接子包含甲基化胞嘧啶以防止em-seq转化并保持ngs文库制备相容性),并进行em-seq转化程序,由此未修饰的胞嘧啶(而不是mc和hmc)经历脱氨基。在这样的脱氨基之后,将分区准备用于测序并进行全基因组测序。对每个分区单独测序,尽管在可选的程序中可以对分区差异性加标签(例如,在分区之后和em-seq转化之前,或者在分区和em-seq转化之后和进一步测序准备之前)、汇集、处理和并行测序。
[0561]
来自高甲基化可变靶区的序列数据是生物信息学上分离的,尽管在可选的程序中,靶区可以在测序前在体外富集。对高甲基化可变靶区的每碱基甲基化进行定量,如图5中示出的,它示出了来自高甲基化分区的高甲基化可变靶区中每个分子的甲基化cpg数目。x轴指示每个分子的cpg总数,因此沿对角线的点代表每个cpg处具有甲基化的分子。因此,可以以单碱基分辨率分析甲基化,并定量mbd分区的材料的每碱基甲基化和部分分子甲基化。与来自健康受试者的样品相比,来自患有结肠直肠癌的受试者的样品在这些区域中表现出高得多的总体甲基化。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。