异噁唑草酮的制备方法与流程
- 国知局
- 2024-06-20 11:06:17
本发明涉及有机合成,具体涉及一种异噁唑草酮的制备方法。
背景技术:
1、异噁唑草酮(isoxaflutole)是罗纳-普朗克公司开发的一类对羟基苯基丙酮酸酯双氧化酶(hppd)抑制剂,具有广谱的除草活性,可通过植物根系或叶面吸收,苗前和苗后均可使用。主要用于玉米田、甘蔗、甜菜等旱田防除多种一年生阔叶及禾本科杂草。
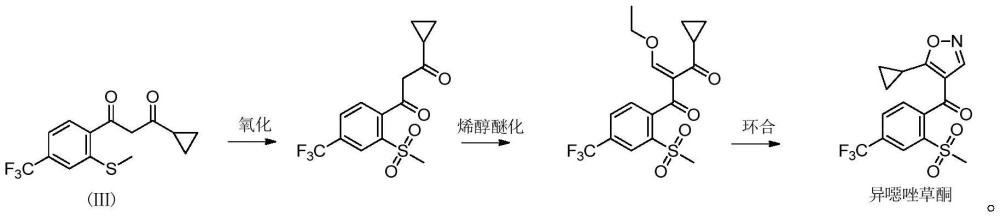
2、罗纳-普朗克公司开发出的异噁唑草酮合成工艺为先氧化,再烯醇醚化、环合(ep0496630b1),其合成路线如下:
3、
4、但是,甲砜基是一种易离去基团,在后续的烯醇醚化、环合步骤中会生成脱甲砜基杂质,故us6392099b1报道了先烯醇醚化、环合,后氧化的如下合成路线:
5、
6、其中,式(ⅲ)所示化合物与原甲酸三乙酯和乙酸酐进行烯醇醚化反应,再与羟胺的无机酸盐在有缚酸剂存在的条件下进行环合反应,氧化步骤采用间氯过氧苯甲酸作氧化剂。
7、但是,该合成路线存在如下问题:间氯过氧苯甲酸价格较高,且副产物难以处理;缚酸剂会生成大量废盐,环境不友好;对于烯醇醚化步骤,乙酸酐成本较高,且会生成乙酰化副产物杂质:
8、
9、cn110128308b报道了一种羧酸作溶剂,路易斯酸催化下,原甲酸三乙酯作为烯醇醚化试剂的方法,避免了使用乙酸酐。但该方法需将溶剂切换为羧酸(文中强调了羧酸类物质作为溶剂的必要性),操作上较为繁琐,存在溶剂交叉问题,且需要额外的溶剂回收设备及能耗。另外,羧酸类溶剂本身对设备腐蚀性较大。
10、对于式(ⅲ)所示化合物的合成,罗纳-普朗克公司报道了两种方法,一是通过酮酯缩合反应(ep0994840b1):
11、
12、该方法的问题在于两个原料缩合反应需在强碱性条件下进行,但在强碱性条件下原料甲酯会发生水解的副反应,大大降低了收率。另外,原料甲酯需由对应的羧酸先氯化再酯化制备,成本较高。
13、另一种方法是环丙基甲酮在强碱性条件下对式(ⅰ)所示化合物中的氰基进行加成反应,得到式(ⅱ)所示化合物,再在酸性条件下水解(ep0705243b1):
14、
15、该方法的问题同上类似,两个原料加成反应需在强碱性条件下进行,但在强碱性条件下,式(ⅰ)所示化合物中的氰基会发生水解的副反应。
16、对于(ⅰ)所示化合物的合成,罗纳-普朗克公司报道了以3-硝基-4-氰基三氟甲基苯或3-氯-4-氰基三氟甲基苯为起始原料,与甲硫醇的金属盐反应的工艺(ep0994852b1)。关于溶剂的选用,尽管提及了芳香烃及卤代烷等与水不互溶体系添加相转移催化剂也可进行反应,但文中强调了酮类、醚类、酰胺类、砜类等大极性溶剂是更优选择,而这类溶剂通常回收困难。
17、
18、综上,现有的各步合成工艺皆存在一些问题,急需一种新的合成工艺以满足工业化生产的需要。
技术实现思路
1、本发明的目的是为了克服现有技术存在的上述问题,提供一种异噁唑草酮的制备方法,该方法简化了工艺操作、抑制了一些杂质的生成,并且具有高反应选择性及转化率。
2、为了实现上述目的,本发明提供一种异噁唑草酮的制备方法,该制备方法包括以下步骤:
3、(1)甲硫化反应:在第一相转移催化剂、有机溶剂和水的存在下,使3-硝基-4-氰基三氟甲基苯和/或3-氯-4-氰基三氟甲基苯与甲硫醇盐进行甲硫化反应,然后静置分层,得到含有式ⅰ所示化合物的有机相;
4、(2)加成反应:在醇盐的存在下,使步骤(1)得到的有机相中的式ⅰ所示化合物与环丙基甲酮进行加成反应,并采出轻组分,反应完成后在反应产物中加入酸进行酸化处理,然后静置分层,得到含有式ⅱ所示化合物的有机相;
5、(3)水解反应:在第二相转移催化剂和酸的存在下,使步骤(2)得到的有机相中的式ⅱ所示化合物水解,然后静置分层,得到含有式ⅲ所示化合物的有机相;
6、(4)烯醇醚化反应:在路易斯酸的存在下,使步骤(3)得到的有机相中的式ⅲ所示化合物与原甲酸三乙酯进行烯醇醚化反应,并采出轻组分,得到含有式ⅳ所示化合物的有机相;
7、(5)环合反应:在第三相转移催化剂、羟胺盐和水的存在下,使步骤(4)得到的有机相中的式ⅳ所示化合物进行环合反应,然后静置分层,得到含有式ⅴ所示化合物的有机相;
8、(6)氧化反应:在氧化催化剂和第四相转移催化剂的存在下,用过氧化氢氧化步骤(5)得到的有机相中的式ⅴ所示化合物,然后静置分层,得到含有异噁唑草酮的有机相;
9、其中,所述有机溶剂为甲苯和/或氯苯;
10、
11、优选地,步骤(1)中,所述第一相转移催化剂为四丁基溴化铵、苄基三甲基氯化铵和苄基三乙基氯化铵中的一种或多种,优选为四丁基溴化铵;更优选地,相对于1摩尔的3-硝基-4-氰基三氟甲基苯和/或3-氯-4-氰基三氟甲基苯,所述第一转移催化剂的用量为0.001~0.1摩尔,优选为0.005~0.02摩尔。
12、优选地,步骤(1)中,所述甲硫醇盐为钠盐、钾盐、锂盐中的一种或多种,优选为钠盐;更优选地,相对于1摩尔的3-硝基-4-氰基三氟甲基苯和/或3-氯-4-氰基三氟甲基苯,所述甲硫醇盐的用量为1~5摩尔,优选为1.15~1.25摩尔。
13、优选地,步骤(1)中,以3-硝基-4-氰基三氟甲基苯作为起始原料时,甲硫化反应的温度为0~50℃,优选为20~30℃;以3-氯-4-氰基三氟甲基苯作为起始原料时,甲硫化反应的温度为50~100℃,优选为70~80℃。
14、优选地,步骤(2)中,所述醇盐为异丙醇、异丁醇、仲丁醇和叔丁醇中的一种或多种的碱金属盐,优选为叔丁醇钠;更优选地,相对于1摩尔的式ⅰ所示化合物,所述醇盐的用量为1~5摩尔,优选为1.4~1.6摩尔。
15、优选地,步骤(2)中,相对于1摩尔的式ⅰ所示化合物,环丙基甲酮的用量为1~5摩尔,优选为1.2~1.4摩尔。
16、优选地,步骤(2)中,加成反应的条件包括:温度为40~110℃,优选为55~65℃,压力为-0.095mpa~常压,优选为-0.095mpa~-0.05mpa。
17、优选地,步骤(2)中,酸化处理中所用的酸为盐酸、硫酸和磷酸中的一种或多种,优选为硫酸;更优选地,酸化处理的终点ph值1~6,优选为4~6。
18、优选地,步骤(2)中,所述加成反应的方法包括:滴加环丙基甲酮的同时,负压采出轻组分。
19、优选地,步骤(3)中,所述第二相转移催化剂为四丁基溴化铵、苄基三甲基氯化铵和苄基三乙基氯化铵中的一种或多种,优选为四丁基溴化铵;优选地,相对于1摩尔的式ⅱ所示化合物,所述第二相转移催化剂用量为0.001~0.5摩尔,优选为0.01~0.05摩尔。
20、优选地,步骤(3)中,水解反应所用的酸为盐酸、硫酸和磷酸中的一种或多种,优选为硫酸;更优选地,相对于1摩尔的式ⅱ所示化合物,酸的用量为1~5摩尔,优选为1.6~1.8摩尔;更优选地,水解反应所用的酸的浓度为30~90重量%,优选为55~65重量%。
21、优选地,步骤(3)中,水解反应的温度为60~105℃,优选为90~95℃或100~105℃。
22、优选地,步骤(4)中,所述路易斯酸为氯化锌、氯化铝、氯化锡、氯化铁、氯化铜、氯化锑、硫酸镍和三氟甲磺酸钠中的一种或多种,优选为氯化锌;更优选地,相对于1摩尔的式ⅲ所示化合物,所述路易斯酸的用量为0.01~1摩尔,优选为0.05~0.2摩尔。
23、优选地,步骤(4)中,相对于1摩尔的式ⅲ所示化合物,原甲酸三乙酯的用量为1~5摩尔,优选为1.5~2.5摩尔。
24、优选地,步骤(4)中,烯醇醚化反应的条件包括:温度50~100℃,优选为50~60℃,压力为-0.095mpa~常压,优选为-0.095~-0.085mpa。
25、优选地,步骤(4)中,烯醇醚化反应的体系中不含有羧酸。
26、优选地,步骤(5)中,所述羟胺盐为硫酸羟胺和/或盐酸羟胺,优选为硫酸羟胺;更优选地,相对于1摩尔的式ⅳ所示化合物,所述羟胺盐的用量为0.5~2摩尔,优选为0.5~0.6摩尔。
27、优选地,步骤(5)中,所述第三相转移催化剂为四丁基溴化铵、苄基三甲基氯化铵和苄基三乙基氯化铵中的一种或多种,优选为四丁基溴化铵;更优选地,相对于1摩尔的式ⅳ所示化合物,所述第三相转移催化剂的用量为0.001~1摩尔,优选为0.01~0.1摩尔。
28、优选地,步骤(5)中,环合反应的温度为30~80℃,优选为40~50℃。
29、优选地,步骤(6)中,所述氧化催化剂为钨酸钠;优选地,相对于1摩尔的式ⅴ所示化合物,所述氧化催化剂的用量为0.001~0.1摩尔,优选为0.001~0.01摩尔。
30、优选地,步骤(6)中,所述第四相转移催化剂为四丁基溴化铵、苄基三甲基氯化铵、苄基三乙基氯化铵中的一种或多种,优选为四丁基溴化铵;更优选地,相对于1摩尔的式ⅴ所示化合物,所述第四相转移催化剂的用量为0.001~0.5摩尔,优选为0.01~0.05摩尔。
31、优选地,步骤(6)中,相对于1摩尔的式ⅴ所示化合物,过氧化氢的用量为2.5~5.0摩尔,优选为3.0~3.5摩尔。
32、优选地,步骤(6)中,氧化反应的温度为10~100℃,优选为35~75℃。
33、优选地,该方法还包括:(7)产品后处理:将步骤(6)得到的含有异噁唑草酮的有机相经脱溶、结晶、过滤、干燥得到异噁唑草酮固体。
34、通过上述技术方案,本发明的合成路线具有如下有益效果:
35、(1)反应溶剂单一,避免了多种溶剂切换带来的交叉污染、溶剂回收设备多、能耗高等问题,大大简化了工艺操作。
36、(2)通过新的工艺条件,有效控制了某些杂质的生成,反应选择性及转化率高,三废量小,适合工业化生产。
37、(3)总收率可以达到约75%以上,产品含量大于98%(w/w)。
本文地址:https://www.jishuxx.com/zhuanli/20240619/1046.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。