一种基于多头注意力机制和图同构神经网络的病毒识别方法
- 国知局
- 2024-08-22 14:23:36
本技术属于深度学习和生物测序,特别是涉及一种基于多头注意力机制和图同构神经网络的病毒识别方法。
背景技术:
1、病毒是地球上最丰富的生物实体之一,广泛存在于土壤、海洋、人体等多种环境中。它们通过控制宿主数量和充当基因转移的媒介,在微生物环境中扮演重要的角色。相比于在实验室培养的病毒发现方法,宏基因组学测序技术可以不用培养,直接从大量样本中提取所有的基因遗传物质。该方法突破了传统方法的限制,快速准确地确定基因的序列信息,极大促进了病毒发现的速度,成为基因测序的主要方法。
2、但正是由于未进行培养,宏基因组学技术提取出的大量序列缺乏分类、宿主等相关信息。在微生物群落研究中,无论是针对水平基因转移探索而研究温和病毒,还是针对噬菌体疗法而研究毒性病毒,将病毒从宏基因组测序数据中准确快速地识别出来是首要前提。因此,病毒识别一直是微生物研究领域的热点。
3、由于毒性病毒和温和病毒都可以将宿主的遗传物质整合到自己的基因组中,导致病毒和宿主基因组之间存在局部序列相似性。这些共同区域给区分病毒和它们的宿主带来了挑战。
4、现有的病毒识别策略包括:
5、1.计算待检测基因序列与现存病毒的相似度,根据相似度推断出待检测基因序列是否为病毒。该策略方法对现有的数据库依赖很大,然而相较于病毒总数,当前数据库中已记载的病毒数量十分有限,无法识别新病毒。待检测基因序列需与现存病毒数据库序列依次比对,比对过程消耗时间巨大,耗时长是阻碍该方法应用的主要原因。微生物群落中存在大量未知病毒,其与当前数据库中的已知病毒相似度较低,所以使用基于相似度的方法无法有效识别新病毒。
6、2.根据病毒的特性,人工设计特征,并根据所设计的特征训练机器学习模型进行病毒识别任务。该策略方法需要人工设计特征,任务量巨大,而且人工设计出特征的质量很大程度上影响病毒识别的准确度,进一步增加了病毒识别任务的难度。
7、3.利用病毒与非病毒数据库训练cnn、lstm等深度学习模型,比如使用卷积神经网络对密码子编码的one-hot编码矩阵进行卷积操作或使用lstm捕捉密码之间的依赖关系,根据所训练的深度学习模型进行病毒识别任务。该策略方法往往仅使用一种模型,仅能捕捉单一维度的特征,无法有效捕捉特征。
技术实现思路
1、为了克服现有技术中的不足,本发明提供一种基于多头注意力机制和图同构神经网络的病毒识别方法,针对宏基因组测序技术所产生的大量基因序列数据,使用多头注意力机制和图同构神经网络训练,使用多头注意力机制和图同构神经网络模型识别病毒序列。
2、为实现上述目的,本发明采用的技术方案如下:
3、一种基于多头注意力机制和图同构神经网络的病毒识别方法,所述方法包括以下步骤:
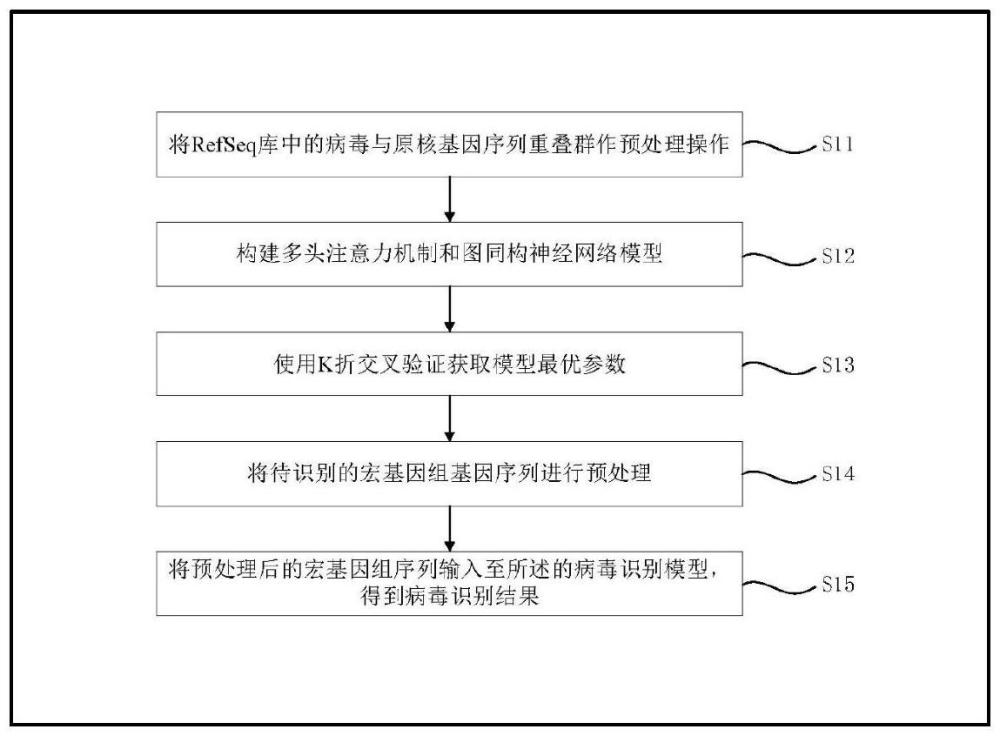
4、步骤1:将refseq库中的病毒与原核基因序列重叠群作预处理操作,用于步骤2模型的训练;
5、步骤2:构建多头注意力机制和图同构神经网络模型;
6、步骤3:使用k折交叉验证获取模型最优参数;
7、步骤4:将待识别的宏基因组基因序列进行预处理;
8、步骤5:将预处理后的宏基因组序列输入至所述的病毒识别模型,得到病毒识别结果。
9、进一步地,所述步骤1具体为:
10、步骤1.1:将refseq库中提取的用于训练的病毒和原核基因序列,过滤出长度大于999bp的基因序列,无重叠地切割为长度为999bp基因序列片段,最后一段长度不满足999bp序列片段舍弃;
11、步骤1.2:将从病毒基因序列切割出的基因序列片段标记为正样本,赋予标签1,将从原核基因序列切割出的基因序列片段标记为负样本,赋予标签0;
12、步骤1.3:将固定长度的基因序列片段,每三个碱基依次进行切割,并依照密码子对应表转化为长度为333的数字序列;
13、步骤1.4:保证测试集中所有测试数据符合实际应用中病毒重叠群第一次被发现的特点,按照病毒重叠群发布日期,将病毒(噬菌体)样本的全基因组划分为训练集和测试集;例如,将2020年之前发布的病毒样本的全基因组划分为训练待训练模型的训练集,将2020年之后发布的病毒样本的全基因组划分为测试待训练模型的测试集。
14、步骤1.5:保证数据平衡(以数量少的病毒为基准,保持病毒和宿主(非病毒)为1:1),避免在由于数据不平衡,导致模型在训练过程中偏向某一类样本的情况;已转换为数字的病毒基因序列片段的数量为基准,随机抽取相同数量的原核基因序列片段,并调整在训练集和训练集中的分布,以使训练数据与测试数据比例按4:1或9:1分布。
15、进一步地,所述步骤2具体为:
16、步骤2.1:模型主要包括嵌入层(embedding)、多头注意力层(multi-headattention)、层归一化层(layernorm)、图同构卷积层(ginconv)、全局累加池化层(global add pooling)、批量归一化层(batchnorm)、全连接层(fc)、随机丢弃层(dropout)、连接层(concat)、激活层对输入数据进行特征提取,多头注意力层捕捉全局依赖关系,得到特征矩阵与权重矩阵,进一步转化为边的邻接矩阵,批量归一化层使神经网络每层的输入都调整为0,方差为1的标准正态分布,激活层使用relu和softmax函数防止模型训练过程中神经元死亡和梯度消失问题的出现,全局累加池化层用于提取多层次的特征,随机丢弃层将训练过程中的神经元按一定比例丢弃,防止过拟合;
17、步骤2.2:使交叉熵损失函数计算预测标签与真实标签之间的损失值,并使用adam优化器优化模型中的参数,使参数能够对输入的基因序列作非线性变换拟合输出,使得损失函数最小化。
18、进一步地,所述步骤3具体为:使用k折交叉验证方法对基于深度学习算法的模型(包括卷积神经网络、多头注意力机制、图同构神经网络)进行评估,k折交叉验证将数据集分成训练集与测试集,在样本数量不充足的情况下,为了充分利用数据集对算法效果的测试,将数据集随机分为k份,每次将其中一份作为测试集,其余k-1份作为训练集进行训练。具体评估的过程为:首先,设置出不同超参数(人为设置调整的参数,比如学习率、生成边的阈值,等等)组合,参数设定可以人为设定10组或者5组,在每组参数上进行k折交叉验证得出平均准确率,(k折交叉验证得到的准确率不具有偶然性),然后将准确率最大的一组超参数作为最优参数。
19、进一步地,k折交叉验证方法使用5折交叉验证获得模型的最终准确率。
20、进一步地,所述步骤4具体为:
21、步骤4.1:从宏基因组测序数据中过滤出长度大于999bp的基因序列,将每一个基因序列,随机起点,固定长度999bp随机切割10个序列片段,序列片段可以有重叠,比如长度为5000的序列,可能随机到9这个位置,然后向后截取999bp的序列片段,可能下次随机到111这个位置,这样随机截取10条;
22、步骤4.2:将序列片段每三个碱基进行切割,并依照密码子对应表转化为长度为333的数字序列。
23、进一步地,步骤5包括以下内容:
24、步骤5.1:将每一个待测基因序列重叠群所切割经过预处理的10个序列片段输入至所述优化后的病毒识别模型,得到10个预测概率;
25、步骤5.2:将每一个基因序列所得到的10个预测概率进行累加取平均操作,最终将概率最大位置的标签作为预测结果,如果预测标签为1,则该基因序列重叠群为病毒,如果预测标签为0,则该基因序列重叠群为非病毒。
26、本发明一种基于多头注意力机制和图同构神经网络的病毒识别方法,利用多头注意力机制捕捉基因序列中密码子之间的全局依赖关系,得到密码子的特征向量矩阵和关系权重矩阵,并根据设定的边的阈值,在相关性大的密码子之间构造得到边的邻接矩阵,将密码子的特征矩阵和边的邻接矩阵输入至图同构网络,利用图同构网络捕捉局部拓扑结构特征,将病毒识别问题转化为图分类任务。该模型使用复合模型捕捉多个维度的基因序列特征,基于病毒数据库中已发现的病毒序列对模型进行训练,最终模型可以根据从已知病毒中捕捉到特征,该特征也分布于未知病毒中,而且由于不存在序列比对的环节,所以该模型不依赖于数据库。因此模型具备识别未知病毒的能力,能够快速准确有效地应对宏基因组测序产生的基因序列片段的病毒识别问题。
本文地址:https://www.jishuxx.com/zhuanli/20240822/278525.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表