一种基于分子动力学模拟配体门控离子通道的路径识别方法
- 国知局
- 2024-10-15 09:35:25
本技术涉及计算机技术、生物大分子多尺度模拟技术及计算结构生物学,具体涉及一种基于分子动力学模拟配体门控离子通道的路径识别方法及装置。
背景技术:
1、配体门控离子通道(ligand-gated ion channels,lgics)是一类在细胞膜上广泛分布的蛋白复合物,在电化学梯度的驱动下,介导带电离子从高浓度一侧流向低浓度一侧。lgics被特定的配体结合后,会引起构象的特定改变,从而形成通道的开放和关闭状态。lgics通过配体结合产生的变构效应来调节通道的开闭状态,其变构通信是空间长程氨基酸内通信促进的生物过程,其中配体结合或者远处位点的氨基酸变化时,会远程影响活性位点,这种影响通过多种传导路径而导致通道的打开或关闭。
2、分子动力学模拟是基于牛顿力学、量子力学和统计力学,利用计算机求解分子体系运动方程的方法,已广泛应用于蛋白质的变构效应研究中,通过对构象运动和运动关联性的分析等来揭示蛋白质体系的变构路径和关键残基。
3、lgics的变构效应通常在毫秒(ms,10-3s)的时间尺度内发生,传统的常规分子动力学模拟(cmd)一般只能捕捉纳秒(ns,10-9s)或微秒(μs,10-6s)的时间尺度的构象变化,因此可能难以捕捉到隐秘的构象变化和变构路径。未结合配体的apo态结构中通常不存在变构口袋,并且只有配体存在时,变构位点处于松弛状态才在构象集合中占主导地位。因此为了捕捉到隐秘的构象变化,需要使lgics先结合上配体。lgics结合上配体后,会诱导周围结构域的构象发生变化,这种构象变化通过多条变构路径传递到孔域,使通道打开。
4、现有技术中结合动力学模拟研究多巴胺受体与配体结合途径,重复29次分子动力学模拟,筛选出在结合途径中起关键作用的残基位点(thomas t,fang y,yuriev e,etal.ligand binding pathways of clozapine and haloperidol in the dopamine d2and d3 receptors[j].journal of chemical information&modeling,2016:acs.jcim.5b00457.)。但lgics结构复杂,仅采用重复的动力学模拟难以捕捉到复杂的变构路径,由于lgics体系庞大,构象变化的时间尺度较大,仅利用传统分子动力学模拟需要消耗大量的人力物力,且其模拟效果难以覆盖所有的构象变化。关于fmrfamide激活的钠通道(fanac),现有技术中结合定点诱变、电生理学以及分子动力学模拟等对其结构和机制研究(fenglian liu,yu dang,lu li,et al.structure and mechanism of aneuropeptide-activated channel in the enac/deg superfamily,nature chemical biology,2023,19(10):1276-1285),筛选出对构象变化起到关键作用的残基,但仅依赖分子动力学模拟并未获得配体与受体的结合变构路径。
5、模拟配体与lgics的结合常采用分子对接技术,常见的分子对接技术包括刚性对接,该方法在计算过程中,参与对接的分子构象不发生变化,仅改变分子空间位置和姿态,方法简单,计算量相对较小,但容易忽略分子的柔性,导致对接结果的准确性降低,且刚性对接无法识别分子在结合过程中可能发生的构象变化,错失一些重要的结合模式,无法真实反映实际的相互作用强度。因此随机结合作为一个重要的前提条件,在研究配体结合产生的变构效应对理解lgics的门控机制和研发新的变构药物具有重要意义。
技术实现思路
1、基于上述内容,本发明提供了一种基于分子动力学模拟配体门控离子通道的路径识别方法或一种配体门控离子通道与配体结合路径以及变构路径的分子动力学模拟方法,通过常规分子动力学、靶向分子动力学和高斯加速分子动力学等多种模拟技术,模拟配体与配体门控离子通道随机结合,获得配体门控离子通道从apo态到holo态的变构路径,使用聚类分析、结合自由能分析、相互作用分析和动态网络分析等分析方法,分析配体进入结合口袋的配体结合路径,进而分析与孔域之间的变构路径。本技术所述方法实现配体与配体门控离子通道的随机结合,可以在较短模拟时间内获取配体门控通道的中间态结构和与孔域之间的变构路径,尤其是识别出比现有技术更多更新的位点,以及确定了最短变构路径,对研究其门控机制和开发新的变构调节剂来调节该通道提供了新的研究思路。
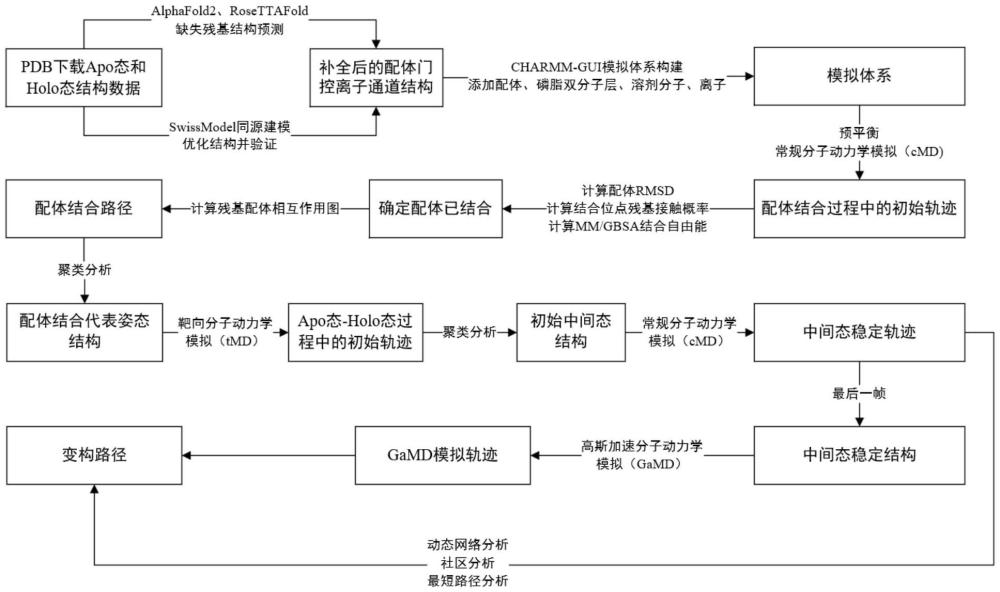
2、本技术的第一方面,提供了一种基于分子动力学模拟配体门控离子通道的路径识别方法,所述的路径识别方法包括:
3、步骤一:获取配体门控离子通道的holo态和apo态的晶体结构作为初始结构,构建模拟体系;
4、步骤二:对模拟体系进行常规分子动力学模拟,获得配体门控离子通道与配体结合的初始轨迹并提取出配体门控离子通道与配体结合的代表姿态结构;
5、步骤三:对配体门控离子通道与配体结合的代表姿态结构和holo态的晶体结构进行靶向分子动力学模拟,提取初始中间态结构;
6、步骤四:对初始中间态结构进行常规分子动力学模拟,获得中间态稳定轨迹并提取中间态稳定结构;
7、步骤五:对中间态稳定结构进行高斯加速分子动力学模拟,获得高斯加速分子动力学模拟轨迹;
8、步骤六:对步骤四获得的中间态稳定轨迹与步骤五获得的高斯加速分子动力学模拟轨迹进行动态网络分析,优选还包括相关系数分析和/或最短路径分析,获得配体与配体门控离子通道结合的变构路径。
9、所述的holo态为结合配体的配体门控离子通道,apo态为未结合配体的配体门控离子通道。
10、所述路径识别方法中常规分子动力学模拟包括模拟配体与配体门控离子通道的随机结合。
11、具体的,步骤一中所述获取配体门控离子通道的holo态和apo态的晶体结构包括:从数据库获取配体门控离子通道的holo态和apo态的晶体结构,删除apo态的晶体结构中除了目标配体门控离子通道以外的其他组分,删除holo态的晶体结构中除了目标配体门控离子通道和配体以外的其他组分;重新构建晶体结构中缺失的结构区域。
12、优选的,所述的数据库包括pdb数据库或rcsb数据库。
13、优选的,所述的重新构建晶体结构中缺失的结构区域包括预测缺失的specificinsertionⅱ区域,然后补全。
14、在本发明的一个具体实施方式中,所述的重新构建晶体结构中缺失的结构区域包括对缺失的氨基酸部分使用alphafold2和rosettafold分别预测结构,并验证最优结构,之后使用swissmodel同源建模进一步优化结构,使用ramachandran图验证结构合理性。
15、具体的,步骤一中所述的模拟体系中包括配体门控离子通道的apo的晶体结构或holo态的晶体结构;所述的模拟体系中还包括配体、磷脂双分子层、溶剂分子和离子。
16、优选的,配体门控离子通道与配体的距离为以上,例如15、16、17、18、19、及以上。
17、进一步优选的,配体门控离子通道的胞外结构域与配体的距离为以上,例如15、16、17、18、19、及以上。
18、在本发明的一个具体实施方式中,配体与配体门控离子通道或其胞外结构域以不低于的距离呈正方形均匀排布,以保证配体的随机结合过程。
19、优选的,模拟体系中原子总数为200000-400000个,例如200000、250000、300000、350000、400000个。
20、在本发明的一个具体实施方式中,所述的步骤一具体包括:
21、步骤1.1:从pdb数据库获取配体门控离子通道的holo态和apo态的晶体结构;
22、步骤1.2:删除apo态的晶体结构中除了目标配体门控离子通道以外的其他组分,删除holo态的晶体结构中除了目标配体门控离子通道和配体以外的其他组分;重新构建晶体结构中缺失的结构区域;
23、步骤1.3:在配体门控离子通道的apo态晶体结构中随机添加配体;
24、步骤1.4:在charmm-gui软件中构建模拟体系,所述的模拟体系中包括配体门控离子通道的apo态的晶体结构或holo态的晶体结构;所述的模拟体系中还包括配体、磷脂双分子层、溶剂分子和离子。
25、优选的,步骤1.4中所述的溶剂分子包括水分子。
26、所述的步骤二具体包括:
27、步骤2.1:对模拟体系进行能量最小化和加热,在npt系综下进行无约束的常规分子动力学模拟,优选将模拟体系运行到一个相对平衡的状态;
28、步骤2.2:计算配体重原子的均方根偏差、配体与结合位点残基接触概率和配体与配体门控离子通道的结合自由能,优选还包括残基与配体相互作用,确定配体结合位点和/或结合路径;
29、步骤2.3:对配体门控离子通道与配体结合的初始轨迹进行聚类分析,提取每个集群的质心结构作为配体门控离子通道与配体结合的代表姿态结构。
30、优选的,步骤2.1所述的对模拟体系进行能量最小化包括通过向配体门控离子通道和脂质添加限制力来实现能量最小化。
31、所述的限制力为0.5-15kcal.mol-1.范围内的任一数值,优选的,所述的限制力为1-10kcal.mol-1.范围内的任一数值,例如0.5、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15kcal.mol-1.
32、在本发明的一个具体实施方式中,通过向配体门控离子通道添加10.0kcal.mol-1.的限制力,向脂质添加5.0kcal.mol-1.的限制力实现能量最小化。
33、优选的,步骤2.1所述的加热包括模拟体系在nvt系综中的温度在100-300ps的时间内上升到300-350k,优选的,所述的加热包括模拟体系的温度在200-300ps的时间内上升到300-320k。
34、在本发明的一个具体实施方式中,所述的加热为模拟体系在nvt系综中的温度在250ps内加热到310k。
35、步骤2.1所述的常规分子动力学模拟在namd2和namd3中执行。
36、所述常规分子动力学模拟的步长为1-2fs,每隔20-100ps保存轨迹,优选的,所述的常规分子动力学模拟的步长为2fs,每隔100ps保存轨迹。
37、步骤2.1所述的常规分子动力学模拟包括将非键相互作用的截止距离为范围内的任一数值,例如优选的,所述的非键相互作用的截止距离为
38、步骤2.1所述的常规分子动力学模拟在npt系综中进行,所述的npt系综的模拟温度为300-310k,压力为1个大气压,采用粒子网格ewald方法处理长程静电相互作用。
39、所述常规分子动力学模拟的时间为0.001-10μs范围内任一数值,优选的,所述的常规分子动力学模拟的时间为0.1-5μs,例如0.001μs、0.1μs、0.5μs、1μs、1.5μs、2μs、2.5μs、3μs、3.5μs、4μs、5μs、6μs、7μs、8μs、9μs、10μs。
40、所述的常规分子动力学模拟的次数为1-5次,例如1、2、3、4、5,优选的,所述的常规分子动力学模拟的次数为3次。
41、在本发明的一个具体实施方式中,所述的常规分子动力学模拟的时间为1μs,模拟3次。
42、步骤2.2所述的接触指配体与结合位点残基之间的重原子距离不大于例如不大于
43、配体的rmsd和结合位点残基的接触概率是基于氨基酸残基的重原子所计算的,当配体rmsd在波动后处于稳定状态可以认为配体可能已经处于结合状态,于是计算与配体产生相互作用的所有残基的配体接触概率,确定重要残基,之后计算残基与配体的相互作用图,按照配体与蛋白有相互作用的时刻作为起始时间,配体稳定结合在蛋白上的口袋中的时刻作为最后时间。
44、所述残基与配体的接触概率不少于0.7时,认为该位点是配体与配体门控离子通道的结合位点。
45、步骤2.2所述的计算配体与配体门控离子通道的结合自由能的方法包括分子动力学/广义玻恩表面积法、热力学积分法或自由能微扰法。
46、残基与配体产生相互作用是残基的所有重原子在配体的所有重原子不大于的距离中,计算残基与配体的相互作用图时,要按照距离的反比归一化相互作用强度。
47、步骤三中所述的靶向分子动力学模拟的起始为配体门控离子通道与配体结合的代表姿态结构,终点为holo态的晶体结构。
48、所述的靶向分子动力学模拟在npt系综中进行。
49、所述的npt系综的模拟温度为300-310k,压力为一个大气压的周期边界条件,其中与h原子相连的键长,限制的阈值为非键相互作用的截断值为
50、所述的步骤三还包括对靶向分子动力学模拟的轨迹进行聚类分析,获得初始中间态结构。
51、步骤四所述的常规分子动力学模拟在namd2和namd3中执行。
52、所述的步骤四在进行常规分子动力学模拟之前需要对模拟体系进行能量最小化和加热。
53、所述的对模拟体系进行能量最小化包括通过向配体门控离子通道和脂质添加限制力来实现能量最小化。
54、所述的限制力为0.5-15kcal.mol-1.范围内的任一数值,优选的,所述的限制力为1-10kcal.mol-1.范围内的任一数值,例如0.5、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15kcal.mol-1.
55、所述的加热包括模拟体系在nvt系综中的温度在100-300ps的时间内上升到300-350k,优选的,所述的加热包括模拟体系的温度在200-300ps的时间内上升到300-320k。
56、所述常规分子动力学模拟的步长为1-2fs,每隔20-100ps保存轨迹,优选的,所述的常规分子动力学模拟的步长为2fs,每隔100ps保存轨迹。
57、步骤四所述的常规分子动力学模拟包括将非键相互作用的截止距离为范围内的任一数值,例如优选的,所述的非键相互作用的截止距离为
58、步骤四所述的常规分子动力学模拟在npt系综中进行,所述的npt系综的模拟温度为300-310k,压力为1个大气压,采用粒子网格ewald方法处理长程静电相互作用。
59、步骤四所述的常规分子动力学模拟的时间为200-250ns范围内的任一数值,例如200ns、205ns、210ns、215ns、220ns、225ns、230ns、235ns、240ns、245ns、250ns,优选的,所述的常规分子动力学模拟的时间为210ns。
60、步骤四中所述的中间态稳定结构为中间态的稳定轨迹的最后一帧。
61、步骤五所述的高斯加速分子动力学模拟的时间为0.5-2μs范围内的任一数值,例如0.5μs、0.6μs、0.7μs、0.8μs、0.9μs、1μs、1.2μs、1.4μs、1.6μs、1.8μs、2μs,优选的,所述的高斯加速分子动力学模拟的时间为1μs。
62、所述的步骤六具体包括:
63、步骤6.1:将中间态稳定轨迹和高斯加速分子动力学模拟的轨迹整合,间隔提取配体门控离子通道的构象,形成完整轨迹;
64、步骤6.2:将步骤6.1获得的完整轨迹输入networkx软件中,生成动态网络;
65、步骤6.3:计算动态网络中配体门控离子通道残基的cα原子与配体的相关系数分析(例如广义相关系数);其中,按照广义相关系数与距离生成动态网络,使用floyd-warshall算法生成变构路径,解释配体调控配体门控离子通道的变构调节机制;
66、步骤6.4:对动态网络进行社区分析和最短路径分析。
67、优选的,步骤6.2生成的动态网络中,配体门控离子通道残基的cα原子和配体关键原子用节点表示,如果两个节点的重原子的距离在75%的采样时间内都在以内,则在两个节点之间加一条边。
68、动态网络中每条边的厚度按距离缩放,边越厚表示两个节点的相关性越大。
69、动态网络中两个节点之间的路径长度为节点集之间涉及的单独路径长度的总和。
70、动态网络中两个节点之间最短路径中涉及的配体门控离子通道的残基被认为是对配体门控离子通道变构调节起重要作用的残基。
71、社区分析:使用以广义相关系数为边权的多级算法将动态网络进一步划分为多个不同的子网络,这些子网络可能代表着该通道各结构域的通信网络的分布情况,并使用pymol对社区网络可视化分析。
72、变构路径分析:使用floyd-warshall算法搜索网络中变构位点和活性位点两个节点之间距离最短的路径,该路径往往是最可能的或生物学相关的信号传递路径,即该通道蛋白功能结构域之间的变构通讯路径。
73、所述聚类分析采用dbscan算法,基于氨基酸残基的backbone原子进行聚类分析。
74、所述的配体门控离子通道包括三聚体配体门控离子通道、四聚体配体门控离子通道或五聚体配体门控离子通道。
75、所述的配体门控离子通道包括乙酰胆碱受体(nachr)、γ-氨基丁酸受体(gaba受体)、甘氨酸受体(glyrs)、谷氨酸受体(iglur)或退化蛋白/上皮钠通道家族(deg/enac),优选的,所述的退化蛋白/上皮钠通道家族包括酸敏感离子通道(asic)、胆汁酸敏感型离子通道(basic)、水螅钠通道(hynac)或fmrf酰胺激活钠通道(fanac)。
76、在本发明的一个具体实施方式中,所述的配体门控离子通道为fmrf酰胺激活钠通道(fanac),所述fanac的配体为fmrf酰胺;
77、fanac与fmrf酰胺结合的位点包括i147、l149、i152、y156、l168、q172、m182、n183、i185、f188、t271、y272、g273、v274、f453和y454。
78、所述的fmrf酰胺与fanac结合过程中结合自由能低于-0.5的残基包括i147、i152、l168、q172、m179、m182、n183、f188、t271、v274。
79、所述的fmrf酰胺与fanac结合过程中配体与残基接触概率不小于0.7的位点包括i147、l149、i152和n183。
80、fmrf酰胺与fanac蛋白在200ns之后相互作用较强的位点包括v274、y272、t271、n183、q172。
81、所述的fmrf酰胺与fanac结合的最短变构路径为n183-f181-s367-l353-n345-c435-y329、v274-n183-i147-e263-q525-e518或f453-q455-r461-h209-l203-k197-i185-n191-w387。
82、所述的fmrf酰胺与fanac结合过程中的关键位点包括q172、n183、v272、f453和l563。
83、本发明的第二方面,提供了一种配体门控离子通道与配体结合路径以及变构路径的分子动力学模拟方法,所述的分子动力学模拟方法包括第一方面所述的路径识别方法。
84、本发明的第三方面,提供了一种基于分子动力学模拟配体门控离子通道的路径识别装置,所述的装置包括存储模块,所述的存储模块包括实现第一方面所述的路径识别方法的程序和/或模型。
85、本发明的第四方面,提供了一种上述第一方面所述的路径识别方法或第二方面所述的分子动力学模拟方法或第三方面所述的路径识别装置在模拟配体门控离子通道与配体结合路径及变构路径的应用。
86、本发明的第五方面,提供了一种上述第一方面所述的路径识别方法或第二方面所述的分子动力学模拟方法或第三方面所述的路径识别装置在筛选与配体门控离子通道结合的变构调节剂中的应用。
87、所述的变构调节剂包括变构激活剂或变构抑制剂。
88、有益效果:
89、(1)本发明主要针对的对象是配体门控离子通道,离子通道作为最复杂的膜蛋白之一,它具有体系更大更复杂、构象变化较小等特点,因此其配体结合路径与变构路径也是较为复杂的,其对于其他蛋白具有更大的参考意义,相比现有技术公开的蛋白,本发明的研究方案具有更好的可扩展性和延伸性。
90、(2)本发明利用分子动力学模拟配体与配体门控离子通道随机结合的过程,分子动力学模拟是分子模拟中最接近实验条件的模拟方法,能够从原子层面模拟出体系的微观演化过程,与常规的刚性对接相比,本发明的方法准确性更高,注重结合过程中配体门控离子通道的构象变化,获得的变构路径更加真实有效。
91、(3)本发明借助分子动力学模拟、接触概率与配体残基相互作用分析、自由能分析和动态网络分析,识别了lgics的配体结合路径、提取出lgics在apo态和holo态之间的重要中间态结构和分析其变构路径。以fmrf酰胺门控钠通道(fanac)为例,成功识别了fmrf酰胺进入结合口袋的结合路径及其结合位点,结合位点包括i147、l149、i152、y156、l168、q172、m182、n183、i185、f188、t271、y272、g273、v274、f453和y454,较现有技术,发现更多的结合位点。提取出apo态和holo态之间的中间态结构,本发明同样适用于其他配体门控通道的研究。
92、(4)本发明使用了常规分子动力学、靶向分子动力学和高斯加速分子动力学等多种模拟方法,保证了配体的随机结合过程的同时使提取出的重要中间态构象更加稳定和可信,又克服传统分子动力学模拟的时间尺度限制,降低了算力消耗。
93、(5)本发明整合了包括动态网络分析等多种分析方法用于揭示变构路径、中间态结构、配体结合路径,动态网络分析可以分析变构信息的传递效率,识别对变构信息传递起重要作用的变构路径及其重要残基,辅助lgics的门控机制研究和重要变构调节剂的研发。以fmrf酰胺门控钠通道(fanac)为例,以识别到的结合位点为变构起点,分别获得多种可能的变构路径,如以n183为变构起点的变构路径包括n183-f181-s367-l353-n345-c435-y329,以v274为变构起点的变构路径包括v274-n183-i147-e263-q525-e518,以f453为变构起点的变构路径包括f453-q455-r461-h209-l203-k197-i185-n191-w387。
本文地址:https://www.jishuxx.com/zhuanli/20241015/314640.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表