一种基于多模态对比学习的P-糖蛋白抑制剂和底物预测方法
- 国知局
- 2025-01-10 13:35:58
本发明涉及深度学习预测药物性质领域,具体涉及一种基于多模态对比学习的p-糖蛋白抑制剂和底物预测方法。
背景技术:
1、p-糖蛋白是一种位于细胞膜上的跨膜蛋白,在肝、肾、小肠、血脑屏障等多种组织中广泛表达,参与药物的吸收、分布、代谢和排泄等过程。它负责将许多有害物质从细胞内排出到细胞外空间,但它也将许多药物推出细胞,其可显著限制或破坏许多药物的活性。鉴于p-gp在药物外排和多药耐药中的关键作用,研究p-gp抑制剂和底物的预测具有重要意义。p-gp抑制剂或底物的活性评估可以通过体内或体外实验获得,然而这些方法具有周期较长、成本高、实验条件不稳定等缺点。因此,迫切需要研发计算方法来辅助p-gp抑制剂和底物的实验筛选。
2、近年来,随着人工智能和化学信息学和发展,机器学习和深度学习方法被越来越多地应用于p-gp抑制剂和底物的预测。然而当前的研究中面临几个显著的挑战。首要挑战在于数据瓶颈,p-gp抑制剂和底物相关的研究数据由于实验环境标准难以统一、存在噪音大等特性,导致现有数据集的质量参差不齐且规模相对有限,这一现状直接限制了预测模型的准确性与泛化能力。
3、此外,在分子表征方面,大多数现有模型依赖于单一的分子表征类型,难以全面、深入地捕捉分子内部复杂多变的结构信息与动态交互过程,从而限制了模型对分子特性的全面理解与精准预测。模型的可解释性缺失也是不容忽视的问题。虽然深度学习等技术在预测精度上屡创佳绩,但其“黑箱”特性使得预测结果背后的生物化学机制变得难以捉摸。这不仅阻碍了对药物作用机制的深入理解,也限制了研究成果在药物研发等实际应用中的转化效率。因此,为推动p-gp抑制剂及底物预测研究的进一步发展,亟需构建高质量、大规模的数据集,探索多模态的分子表征方法以增强预测模型的精度,并加强模型可解释性研究,识别与p-gp相互作用密切相关的关键官能团,为药物研发提供更加坚实可靠的理论支撑与技术支持。
技术实现思路
1、为解决上述问题,本发明提供了一种基于多模态对比学习的p-糖蛋白抑制剂和底物预测方法,构建高质量的p-糖蛋白抑制剂和底物数据集,提升了模型预测精度和泛化性能,并实现了模型的可解释性,为药物开发中的分子设计提供可靠的科学依据。
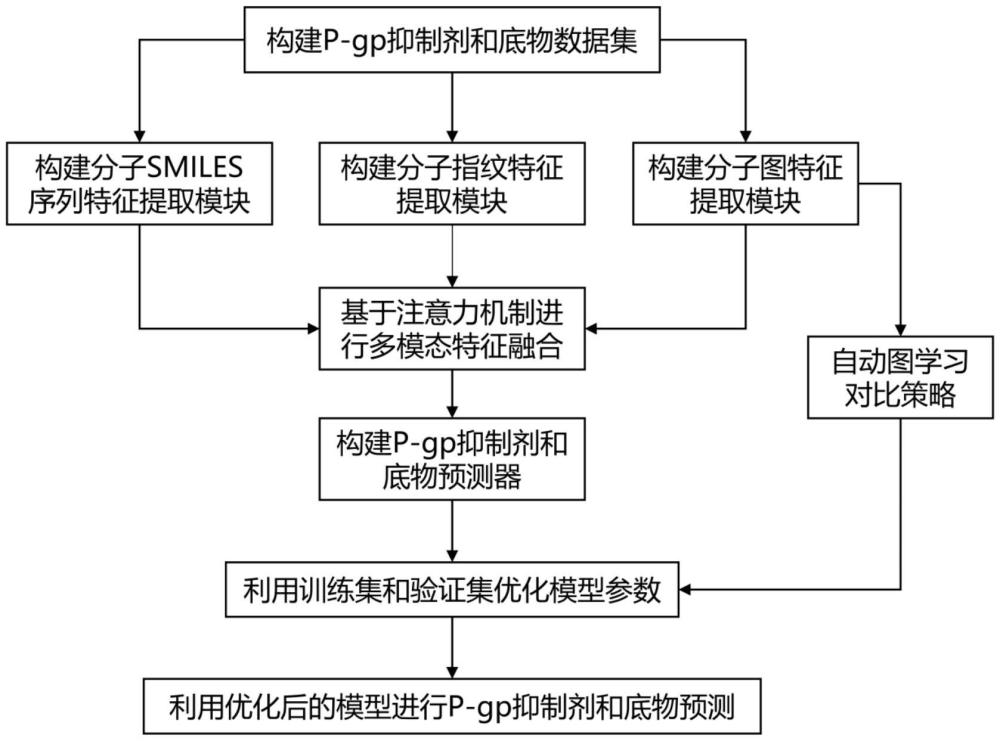
2、一种基于多模态对比学习的p-糖蛋白抑制剂和底物预测方法,包括以下步骤:
3、1)构建p-糖蛋白抑制剂和底物的数据集;
4、2)利用深度学习方法构建p-糖蛋白抑制剂和底物预测模型;
5、3)将步骤1)获取的数据集输入到步骤2)构建的p-糖蛋白抑制剂和底物预测模型中,针对分子图特征引入图对比学习策略,得到数据增广后的分子图,计算分子图和数据增广后的分子图之间的对比损失,根据p-糖蛋白抑制剂和底物预测器输出的融合特征计算分类损失,将对比损失和分类损失联合得到总损失函数,通过总损失函数训练并优化p-糖蛋白抑制剂和底物预测模型的参数,得到优化后的p-糖蛋白抑制剂和底物预测模型;
6、4)将待预测的化合物数据输入到优化后的p-糖蛋白抑制剂和底物预测模型,输出最终的分类结果,判断化合物是否为p-糖蛋白抑制剂和底物。
7、步骤1)中,构建p-糖蛋白抑制剂和底物的数据集,具体包括:
8、从相关数据库中收集分子化合物与p-糖蛋白抑制剂和底物相关的实验数据、化合物smiles序列,整合后进行预处理,获得p-糖蛋白抑制剂和底物的数据集。
9、步骤2)中,所述的p-糖蛋白抑制剂和底物预测模型,具体包括:
10、分子smiles序列特征提取模块,用于接收p-糖蛋白抑制剂和底物的数据集并提取分子smiles序列特征;
11、分子指纹特征提取模块,用于接收p-糖蛋白抑制剂和底物的数据集并生成分子指纹特征;
12、分子图特征提取模块,用于接收p-糖蛋白抑制剂和底物的数据集并生成分子图特征;
13、基于注意力机制构建的特征融合模块,用于接收所述分子smiles序列特征提取模块输出的分子smiles序列特征、所述分子指纹特征提取模块输出的分子指纹特征、所述分子图特征提取模块输出的分子图特征,并将分子smiles序列特征、分子指纹特征和分子图特征进行特征融合输出融合特征;
14、p-糖蛋白抑制剂和底物预测器,包括全连接层,用于接收融合特征并预测输出分类结果。
15、步骤2)中,提取分子smiles序列特征,具体步骤包括:
16、2-1-1)将p-糖蛋白抑制剂和底物的数据集中的smiles序列划分为单个原子或符号,然后从预训练字典中查找相对应的原子的嵌入向量,如果不在字典中,则生成随机嵌入向量;将原子的嵌入向量和随机嵌入向量聚合得到smiles序列的初步表示;
17、2-1-2)通过双向长短记忆网络bi-lstm对smiles序列的初步表示进行预处理,捕获smiles上下文信息表示;
18、2-1-3)通过注意力机制计算smiles上下文信息表示中的每个原子的注意力权重,并根据注意力权重对每个原子的隐藏状态加权求和得到分子smiles序列的特征向量;
19、2-1-4)将分子smiles序列的特征向量传入全连接层得到分子i的smiles序列特征si。
20、步骤2)中,生成分子指纹特征,具体包括:
21、分别计算p-糖蛋白抑制剂和底物的数据集中分子的圆形拓扑指纹ecfp4指纹和pubchem指纹,将两种指纹进行连接得到混合指纹,并将混合指纹传入全连接层,得到分子i的分子指纹特征fi。
22、步骤2)中,生成分子图特征,具体包括:
23、2-3-1)根据p-糖蛋白抑制剂和底物的数据集中的分子smiles提取节点和边信息,使用节点和边的信息构建图的邻接矩阵;
24、2-3-2)计算p-糖蛋白抑制剂和底物的数据集中的每个原子的理化性质,作为分子图中点的初始特征向量;
25、2-3-3)采用图同构网络gin,通过聚合图的邻接矩阵中节点的自身特征和邻居节点的特征混合来更新步骤2-3-2)获得的初始特征向量,通过对所有节点的特征进行全局平均池化操作,得到整个分子的全局图表征;
26、2-3-4)将全局图表征输入到全连接层,得到分子i的分子图特征gi。
27、步骤3)中,总损失函数,具体包括:
28、通过全连接层对融合特征的分子表征mi进行处理,获得用于分类任务的最终分子表征yi,随后计算二元交叉熵损失作为分类损失其计算公式为:
29、
30、其中,n是分子的总数,yi是第i个分子的实际标签,是模型预测第i个分子为正类的概率;
31、针对分子图特征引入图对比学习策略,得到数据增广后的分子图,计算分子图和数据增广后的分子图之间的nt-xent损失作为对比损失
32、最终总损失函数定义为分类损失和对比学习损失的加权和:
33、
34、其中,β为可调整的权重参数。
35、具体地,本发明的技术方案如下:
36、1)构建p-糖蛋白(p-glycoprotein,p-gp)抑制剂和底物数据集,包括从相关数据库中收集小分子化合物与p-糖蛋白抑制剂和底物相关的实验数据、化合物简化分子线性输入规范格式smiles序列等信息,整合后进行预处理,获得最终的数据集;
37、2)利用深度学习方法构建p-糖蛋白抑制剂和底物预测模型,该模型架构包括如下组件:分子smiles序列特征提取模块、分子指纹特征提取模块、分子图特征提取模块、基于注意力机制构建的特征融合模块和基于全连接层构建的预测器;
38、3)在训练过程中,针对分子图特征引入图对比学习策略,得到数据增广后的分子图,计算分子图和增广图之间的对比损失;
39、4)采用步骤1)获取的数据集,并通过计算步骤2)中预测器的分类损失和步骤3)中的对比损失,训练并优化p-糖蛋白抑制剂和底物预测模型的参数,得到优化后的预测模型;
40、5)将待预测的化合物数据输入优化后的p-糖蛋白抑制剂和底物预测模型,输出最终的分类结果,判断化合物是否为p-糖蛋白抑制剂和底物;
41、6)对于预测模型进行可解释性分析,识别化合物中与p-gp相互作用密切相关的关键官能团。
42、步骤1)中,为了构建p-gp抑制剂和底物数据集,具体包括以下步骤:
43、1-1)采用文本挖掘和手工核对方法从数据库和文献中收集与p-gp相关的实验数据,数据库包括chembl、ochem、pubchem、drugbank等;
44、1-2)仔细检查实验数据,依据指定阈值对化合物进行阴阳性标注;
45、1-3)从数据集中删除重复以及分类矛盾的数据;
46、1-4)所有化合物结构进行标准化预处理,包括去除无机物、络合物,将盐转化为相应的酸或碱,对于立体异构体仅保留一条数据等;
47、1-5)额外收集相关研究和数据库的化合物数据作为外部测试集,将以上的数据预处理步骤应用于外部测试集,并删除外部测试集中和数据集重复的化合物和相应生物活性数据。
48、所述步骤2)中,根据输入数据的smiles,通过分子smiles序列特征提取模块提取分子smiles序列特征,具体包括以下步骤:
49、2-1-1)将给定的smiles序列划分为单个原子或符号,然后从预训练字典中查找相对应的原子的嵌入向量,如果不在字典中,则生成随机嵌入向量;预训练字典包含常见原子的嵌入向量;
50、2-1-2)将嵌入向量聚合得到嵌入矩阵其中m是原子数量,d是嵌入向量的维度;
51、2-1-3)通过以上步骤可得到smiles序列的特征向量x={x1,x2,…,xn},其中是第t个原子的特征向量。通过双向长短记忆网络bi-lstm捕获smiles上下文信息表示,得到原子t的隐藏状态ht:
52、
53、其中和分别表示前向和后向的lstm单元,ht-1为原子t-1的隐藏状态;
54、2-1-4)采用注意力机制进一步提升模型对重要序列信息的捕捉能力,采用以下公式计算ht的注意力权重:
55、et=tanh(w1ht+b1) (2)
56、
57、其中et为原子t注意力分数,通过tanh激活函数前馈神经网络进行计算,w1为权重矩阵,b1为偏置向量;对注意力分数采用softmax函数进行归一化处理,得到原子t的注意力权重αt;
58、2-1-5)根据注意力权重对每个原子的隐藏状态加权求和得到分子smiles序列的特征向量将特征向量hs传入全连接层,得到分子i的smiles序列特征si。
59、所述步骤2)中,根据输入数据的smiles,通过分子指纹特征提取模块生成分子指纹特征,具体包括以下步骤:
60、分别计算分子的圆形拓扑指纹ecfp4指纹和基于子结构的pubchem指纹,将两种指纹进行连接得到混合指纹,并将混合指纹传入两层全连接层,得到分子i的分子指纹特征fi。
61、所述步骤2)中,根据输入数据的smiles,通过分子图特征提取模块生成分子图特征,具体包括以下步骤:
62、2-3-1)根据分子smiles提取节点和边信息,其中分子中的每个原子映射为分子图中的节点,原子之间的化学键映射为分子图中的边,使用节点和边的信息构建图的邻接矩阵,以表示节点之间的连接关系;
63、2-3-2)计算每个原子的理化性质,作为分子图中点的初始特征向量;理化性质具体包括原子类型、带电荷数、手性碳情况、相连氢原子数、杂化类型、是否是芳香原子等;
64、2-3-3)采用图同构网络gin,通过聚合邻居节点的特征并与自身特征混合来更新节点表示,第k层gin的节点特征更新规则如下:
65、
66、对所有节点特征进行全局平均池化操作,得到整个分子的全局图表征hg:
67、
68、其中表示节点v在第k层的特征向量,表示节点v的邻居节点集合,mlp(k)表示第k层的多层感知机(mlp),∈是一个可学习的或固定的标量,k表示gin的层数,v表示节点的集合,global_mean_pool代表全局平均池化操作。
69、2-3-4)将全局图表征输入到全连接层,得到分子i的分子图表征gi。
70、所述步骤2)中,采用注意力机制处理以上步骤得到的多模态特征,具体包括以下步骤:
71、2-4-1)针对三种不同的分子表征si(smiles表征)、fi(分子指纹表征)、gi(分子图表征),通过tanh激活函数前馈神经网络计算注意力得分:
72、es=tanh(wssi+bs) (6)
73、ef=tanh(wffi+bf) (7)
74、eg=tanh(wggi+bg) (8)
75、其中es、ef、eg,ws、wf、wg和bs、bf、bg分别是smiles表征、分子指纹表征、分子图表征的注意力得分、权重矩阵、偏置向量;
76、2-4-2)通过对注意力得分归一化计算每个表征的注意力权重,然后将注意力权重应用于相应的分子表征,进行加权融合得到分子i的表征mi:
77、
78、通过计算得到的分类损失和步骤2-5)得到的对比损失来训练优化模型所述步骤3)中,分子图对比学习策略具体包括以下步骤:
79、3-1)对于给定的分子图,通过gin从节点属性中获得节点嵌入,并利用这些嵌入来预测对该节点的增强方式,即预测该节点应被丢弃、保留还是遮掩。然后,采用gumbel-softmax技巧从预测的分布中采样进行节点操作,获得数据增广后的分子图;
80、3-2)随机抽取一批大小为n的分子图,通过以上步骤进行数据增广,生成相等数量的对应增广分子图。给定的分子图表征gi与增广分子图表征构成一个正样本对,与剩余的2(n-1)个图/增广图的表示构成负样本对。nt-xent用于计算对比学习训练的损失,以最大化正样本对表示之间的一致性,同时最小化负样本对表示之间的一致性,将对比学习损失表示为:
81、
82、其中,τ是温度系数,用于调节相似度的尺度,设置为0.2。1[k≠i]是指示函数,当k≠i时为1,否则为0。表示gi和之间的余弦相似度;
83、3-3)对于输入批次的样本对,对比损失通过对该批次中所有正样本对的对比损失进行平均计算而得到:
84、
85、其中和是一对分子图和数据增广后的分子图之间的对比损失,n是分子的总数;
86、所述步骤4)中,计算模型损失函数具体包括以下步骤:
87、4-1)通过全连接层对融合的分子表征mi进行进一步处理,获得用于分类任务的最终分子表征yi。随后计算二元交叉熵损失(bce损失)作为分类损失其计算公式为:
88、
89、其中,n是分子的总数,gi是第i个分子的实际标签,是模型预测第i个分子为正类的概率;
90、4-2)最终总损失函数定义为分类损失和对比学习损失的加权和:
91、
92、其中,β为可调整的权重参数。
93、所述步骤6)中,可解释性分析具体包括以下步骤:
94、6-1)针对分子smiles序列表征,采用注意力权重可视化技术,分析模型在预测过程中分配给各个smiles字符的注意力权重来识别影响预测的关键因素;
95、6-2)针对分子指纹表征,采用积分梯度法,通过分析模型输入和基准输入之间的路径上的梯度来标识重要的分子指纹位点,积分梯度越高,说明该位点所起的作用越大;
96、6-3)针对分子图特征,应用梯度加权类激活映射(grad-cam)算法,利用卷积层的梯度信息来突出显示图中对模型预测贡献最大的区域。通过可视化模型关注的关键原子和化学键,可以进一步探索这些区域与药物活性之间的关联,为药物设计和优化提供指导。
97、与现有技术相比,本发明具有如下优点:
98、(1)本发明设计的多模态表征学习策略,整合了分子smiles序列特征、分子指纹特征以及分子图特征,能够捕捉到分子结构的多个维度信息,提高了模型对p-gp抑制剂和底物识别的准确性和全面性。相比之下,现有的单一模态或简单组合的技术方案往往难以全面反映分子的复杂性质。
99、(2)本发明引入注意力机制进行多模态特征融合,使得模型能够自动学习并强化关键特征的重要性,同时抑制非关键信息,从而提高了多模态特征提取与融合的效率和准确性。
100、(3)本发明利用自动图对比学习策略生成分子增广图,用于最大化同一分子表示之间的相似性和最小化不同分子表示之间的相似性,不仅丰富了训练数据的多样性,还增强了模型的泛化能力。
101、(4)本发明构建的高质量数据集以及采用的标准化数据处理流程,确保了数据的一致性和可靠性,为模型训练提供了坚实的基础。这种严谨的数据准备方式,是提升模型性能的关键。
102、(5)本发明针对三种特征进行了可解释性分析,准确识别出与p-gp相互作用密切相关的关键官能团,为科研工作者进行先导化合物优化提供科学、可靠的信息,有效提升药物研发的效率。
本文地址:https://www.jishuxx.com/zhuanli/20250110/354134.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。