一种多重PCR靶向测序技术的引物设计方法与流程
- 国知局
- 2025-01-17 12:59:41
本发明涉及生物信息学领域,还涉及分子生物学、微生物学和测序,特别是涉及一种多重pcr靶向测序技术的引物设计方法。
背景技术:
1、随着科技的发展,分子生物学、遗传学、生物信息学和测序技术等领域取得了显著的进步。其中,聚合酶链式反应(polymerase chain reaction,pcr)作为分子生物学的基石技术之一,通过体外扩增特定的dna片段,使得微量的dna样品也能被检测出来;而第二、三代测序技术则可以对大量dna片段进行并行测序,从而实现对整个基因组或转录组的快速测序。多重pcr(multiplex polymerase chain reaction,mpcr)技术是pcr技术的一种拓展,允许在一个pcr体系内同时使用多对引物,针对多个靶标序列进行同步扩增。这种方法相比传统pcr具有高效、高通量且低成本的优势,能满足临床上对多种病原体微生物同步检测的需求,从而大幅提升诊断效率和准确性。然而,在实际应用中,这些技术仍存在一些挑战和局限,主要表现在以下几点:
2、1. 基因组库质量筛选问题:高质量的物种基因组库搭建,是后续选取物种特异区段、设计出理想引物的根基。现有的基因组筛选方法多依赖于基因组组装级别的标签(如representative genome、reference genome、complete genome、chromosome等),缺乏更加细致的筛选标准(如基因组测序时间、宿主、来源地、完整度等),导致基因组库中基因组质量参差不齐。
3、2. 引物的兼容性问题:设计多重pcr引物检测目标病原时,由于临床样本中目标病原的基因组种类多样,因此需要引物本身有很高的兼容性,也就是每个目标病原的引物都可拷贝该病原下序列之间具有些许差异的、多样的基因组。现有方法通常先在物种基因组内寻找高保守度的物种特异片段,再以特异片段作为模板设计引物。但对于基因组较多样的物种,寻找特异片段的过程往往不顺利,需要不断调整基因组库进行多轮特异序列的查找,工作量大的同时还很难得到片段长度能够满足引物设计需求且高覆盖度的特异片段。
4、3. 引物二聚体问题:多重pcr体系中由于引物较多,引物之间的二聚体结合会降低体系的效率。常见方法仅基于结合自由能值进行引物二聚体的筛选和优化,未充分考虑引物二聚体结合区域的碱基长度、二聚体结合区域起点和终点距离两条引物3’端距离等因素对二聚体结合情况的影响。
技术实现思路
1、发明目的:本发明的目的是提供一种多重pcr靶向测序病原体的引物设计方法,以避免在获取种内基因组较多样物种特异序列时的大量重复劳动,并提供理想兼容性。
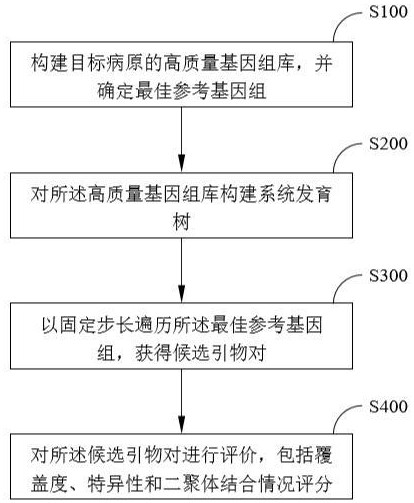
2、技术方案:本发明提供了一种多重pcr靶向测序技术的引物设计方法,所述方法依次包括如下步骤:
3、s100:确定最佳参考基因组,构建目标病原的高质量基因组库;
4、s200:对所述高质量基因组库构建系统发育树;
5、s300:以固定步长遍历所述最佳参考基因组,获得候选引物对;
6、s400:对所述候选引物对进行评价,包括覆盖度、特异性和二聚体结合情况评分。
7、本发明所述的目标病原是指在靶向多重pcr引物体系中,预先设计的引物所靶向的多种生物,包括但不限于病毒、细菌和真菌。
8、对于本发明提供的引物设计及优化的方法适用于高通量分子诊断领域,特别适用于多重pcr对多个目标病原靶标通过一次pcr反应实现检测。由于临床样本中的目标病原基因组存在多样性,选择兼容性高的引物尤为重要,以确保能有效拷贝具有微小序列差异的多样基因组。搭建高质量、多样性的物种基因组库,是保证引物高兼容性的基础。这样在确保引物高度覆盖基因组库的情况下,就可以推断出引物在真实世界中对各种基因组也具有较高的兼容。同时在设计引物时,提高引物对多样基因组的兼容性,并同时采用高效的设计策略减少繁琐的工作量也必不可少,本发明可很好解决这两个难点。
9、本发明构建目标病原的高质量基因组库,区别于根据基因组组装级别顺序进行筛选的现有技术,兼顾了基因组测序时间、宿主、来源地、完整度等多个衡量标准,这样在筛选时可以过滤掉因测序年份过早、测序技术不足而产生的测序质量一般的基因组,或宿主与来源地与中国人群过于远源的基因组,这些基因组若存在于基因组库内,会干扰引物兼容性的评估。同时,为了确保基因组库具有多样性,本发明还使用了构建系统发育树的方法对基因组库进行亲缘关系上的分簇,确保库内基因组既没有大部分集中在某个亚种分支、多样性差,也没有个别远源的进化分支在设计引物时遗漏。高质量基因组库的构建是基于最佳参考基因组,根据组装级别、基因组测序时间、宿主、来源地、完整度等获取其他同物种基因组,且要求同物种基因组的测序时间与当前年份接近,并具有一定样本数量。
10、在设计引物阶段,为提高引物对目标物种的多样基因组的兼容性,业界常见方法是先获取基因组库多个基因组共有的保守片段,然后在这些保守片段上设计引物。为了获取这些高保守片段,需要进行多轮基因组序列比对和聚类,以找出共有片段。这个过程繁琐且耗时,尤其对于一些基因组多样性高的物种,大量工作后依旧未必能顺利找到兼具高保守性和高物种特异性的序列。本发明先在物种基因组库内确定一个最佳参考基因组,再用系统发育树对多样性的物种基因组库进行分簇,若存在远源的簇,则对其进行单独分组,分组中也确认一个最佳参考基因组。在最佳参考基因组上遍历设计候选引物,再计算这些候选引物在基因组库内的覆盖度。对于远源的基因组分组,在分组上的参考基因组上遍历设计候选引物,再计算候选引物在分组内的基因组覆盖度。这样先分组、再设计引物、最后计算引物覆盖度的方式,减少了获取共有片段时,大量的长片段比对工作,更加简洁,也确保了引物的高覆盖度。
11、进一步地,所述步骤s100具体包括如下步骤:
12、s110:获取目标病原的最佳参考基因组;
13、s120:获取最近测序时间的若干其他同物种基因组,所述其他同物种基因组按组装级别由高到低的优先级获取;
14、s130:筛取完整度较高且满足样本数量的其他同物种基因组,当样本数量不足时,优先扩大步骤s120的测序时间。
15、其中,所述最佳参考基因组是基于基因组来源、基因组组装级别、测序时间(年)和checkm值等多个维度进行确定,以确保所筛选的基因组质量高、对物种有代表性。
16、具体来说,所述最佳参考基因组的确定方法为:从refseq数据库或genbank数据库中按照representative genome>reference genome>complete genome>chromosome>scaffold>contig的优先级顺序筛选一个最佳参考基因组;
17、当同级别的基因组或基因组结构有多个时,选取测序年份最近、checkm值最高的基因组或基因组结构;
18、其中,测序年份的优先级大于checkm值;且如果多个同级别基因组或基因组结构的测序年份和checkm值均相同,则任意选择一个。
19、具体来说,refseq数据库中物种通常至少含有representative genome、reference genome的一种基因组。若物种的representative genome或reference genome数量大于1,则选取测序年份距现在最接近、checkm值最高的基因组作为最佳参考基因组,且测序年份的优先级大于checkm值;若测序年份和checkm值均相同,则任意选取一个。若物种在refseq数据库中没有representative genome或reference genome,则在genbank数据库中按上述同样的方式确认最佳参考基因组。若refseq和genbank数据库中都没有representative genome和reference genome,则按照complete genome>chromosome>scaffold>contig的优先级顺序向下进行基因组结构筛选。当同级别的基因组/基因组结构数量大于1,则选取测序年份距现在最接近、checkm值最高的基因组作为最佳参考基因组,且测序年份的优先级大于checkm值;若测序年份和checkm值均相同,则任意选取一个。
20、本发明所述用于确认最佳参考基因组的checkm值是衡量基因组完整度(completeness)的指标。
21、其中,对于分类学上为“种”的病原,所述样本数量不低于10个;对于分类学上为“物种复合体”或“属”的病原,所述样本数量为50个。
22、需要补充说明的是,所述步骤s120优先获取组装级别为complete的基因组样本。当样本数量不足时,优先扩大步骤s120的测序时间以获取更多的样本;当扩大测序时间仍然无法获取足量样本时,再按照组装级别优先级下载chromosome、scaffold、contig等基因直至样本数量达标。
23、作为本发明的进一步优化,对高质量基因组库中的每个目标病原,利用checkm2软件进行完整度检验,筛取完整度大于等于95%的基因组。
24、特别地,部分物种的基因组因构成较为复杂,checkm2无法进行评分,ncbirefseq/genbank的checkm项也均为0,此时则可忽略该项指标。
25、本发明所述的系统发育树又称分子进化树,是通过比较生物大分子(如蛋白质、核酸)序列差异的数值,从而构建的可视化的树图,描述了不同对象之间在序列层面的亲缘关系,是一种生物信息学分析方法。与本技术主题相近的现有技术中,鲜有披露通过构建系统发育树,来检验物种高质量基因组中各个基因组的进化亲缘关系,并应用在多重pcr靶向测序技术的引物设计方法中。本发明基于系统发育树从基因组序列差异性的角度来进行基因组的“分簇”,以体现基因组之间的亲缘关系。具体来说,本发明使用parsnp软件构建系统发育树,它是先将物种内各个基因组的最大相似序列部分进行对齐形成“核心对齐区域”,再在“核心对齐区域”内检测单核苷酸多态性,这些单核苷酸多态性通常被视为研究进化关系最可靠的突变;再利用最大似然法算法,根据“核心对齐区域”的差异性构建进化树,反映这些基因组的亲缘关系。
26、进一步地,所述步骤s200获得的基因组进化树满足如下要求:所述基因组进化树的分支均匀,既没有进化距离明显较远的孤立分支,也没有绝大多数基因组密集分布在同一分支的情况。系统发育树中,叶子结点表示系统发育树的末端节点,在此节点上不再有进一步的分支,本研究中每个叶子节点代表一个参与进化树构建的基因组。初始进化树先对多个序列相近的基因组进行相似性和差异性的分析,形成独立的小型分支结构,多个小型分支结构进而再形成趋向于根节点的分支延伸。分支长度表示两个节点间的亲缘关系,分支越长,亲缘关系越远。进化树分支均匀的判定方式为:在系统发育树中,含有叶子结点最多的小型分支结构里,叶子结点数小于基因组总数的30%~50%。且,若某些进化分支长度与所有进化分支的长度平均值之比大于10~100倍,则这些分支上的基因组视为远源基因组,若分支的基因组个数>总基因组数的0.01-10%,且这些基因组的质量均表现良好,则按进化树分支将这些基因组进行分组,后续进行小组内的引物设计。若分支的基因组个数>总基因组数的0.01-10%,则直接将这些基因组删去。
27、作为本发明的优选方案,是否将明显较远的孤立分支的基因组从高质量基因组库中剔除,以及是否则按进化树分支将超过总基因组数一定比例的进化距离明显较远的基因组进行分组的评价依据为总基因组数的5%。应当理解,在多重pcr引物设计中,为了应对临床样本中目标病原基因组的多样性,需确保引物具备高兼容性,能够覆盖各目标病原的多样基因组序列。因此,需选择测序质量高且普适性的基因组构建高质量基因组库。这要求所选基因组不应集中于单一进化支系(如某个亚种),以免造成普适性不足。同样,当一组基因组在进化关系上过于远源且数量超过总数的5%时,应将其按进化关系划分成若干组,并分别设计引物复盖。
28、对于步骤s300,所述步骤s300中以300 bp为滑动窗口长度,20 bp为滑动步长,进行全基因组的叠瓦式切片,在每个片段上利用primer3软件设计引物。
29、优选地,对于第二代测序技术,扩增子长度为180-300bp,tm值范围55-65℃,引物臂长度为18-25bp,引物臂gc碱基含量为40-60%。
30、对于第三代测序技术,本领域技术人员在上述第二代测序技术的基础上,在不脱离本技术发明原理的前提下,可对滑动窗口长度、滑动步长的设计进行适应性改动,都在本技术的保护范围内。
31、对于步骤s400,所述覆盖度的评价方法是将所述候选引物对双端匹配到所述高质量基因组库的每个其他同物种基因组中,并计算引物覆盖度:
32、引物覆盖度 = 匹配合格的基因组数/基因组总数×100%。
33、其中,匹配合格至少同时满足以下条件:上游引物和下游引物都能实现匹配,引物扩增子的匹配长度不小于扩增子总长度的95%,引物扩增子长度为100 - 300bp。
34、所述特异性的评价方法包括如下步骤:
35、s421:将所述候选引物对在ncbi nt库中成对比对库内所有的基因组;
36、s422:比对后,获得引物插入片段,所述引物插入片段是指在 pcr过程中,由一对互补的寡核苷酸引物所界定并扩增的核酸序列,引物插入片段的起始端由前向引物的3'端定义,结束端由反向引物的5'端定义;
37、s423:将所述引物插入片段输入到kraken 2软件进行验证,从而筛选特异性合格的引物对;
38、s424:当所述引物插入片段所属基因组的物种与引物目标物种一致时,kraken 2的物种判别结果必须是该物种;或引物插入片段所属基因组的物种与引物目标物种不一致时,kraken 2的物种判别结果必须与基因组物种一致。
39、上述方法允许引物与非目标物种的序列结合,同时确保可在kraken 2中根据引物插入片段正确判断非目标物种。这样放宽了对引物特异性的要求,同时也不会因为物种错报而产生假阳,提高了引物的利用率。
40、多重引物体系当中,常见的影响体系扩增效率的原因之一,就是引物之间的二聚体结合,即体系的引物之间因为碱基互补而相互结合,从而使得引物无法与病原的序列结合,降低了引物的扩增效率。业界常见方法是先将体系中所有可能产生结合的二聚体枚举出,并计算这些二聚体结合的自由能,当自由能处在安全阈值范围外(<- 4 kcal/mol),则认为两条引物在真实实验中较容易结合生产二聚体。或是用一些全局最优算法,例如贪婪算法,来进行引物之间的组合,尽量挑选与现有体系内引物二聚体结合最少的新引物加入,循环多次后直至引物数量达标。二聚体结合自由能的计算,有碱基配对、堆叠相互作用、离子浓度、温度指标的参与,它只是衡量了这段碱基在特定温度、溶液浓度下结合的难易程度,却忽视了二聚体的结合情况也与二聚体区域距离两条引物3’端的距离有关,二聚体结合区域距离3’端越近,影响越不好。全局最优算法往往得出的是一套固定的引物组合,当需要对体系中某些引物进行替换时,情况就会很麻烦,或许需要重新进行算法的运行。
41、本发明提出了一种新的引物二聚体评价方法,该方法不仅考虑了本领域技术人员普遍关注的二聚体结合自由能,还综合了引物二聚体结合区域的长度、结合区域gc碱基的数量、以及引物二聚体结合位点与各自引物3'端的距离等因素。传统方法往往依赖于算法按顺序逐个评估引物对,导致结果缺乏灵活性和可控性。本发明提供的评价方法更加直观,允许研究人员自由选择引物对,而不是单纯依赖算法自动生成的固定组合,从而提供了更高的灵活性和选择的自由度。
42、具体来说,所述二聚体结合评分为:
43、;
44、当所述候选引物对中任意一条引物的不低于103时,筛除该引物对应的引物对;
45、“二聚体结合区域”是指在聚合酶链式反应(pcr)过程中,两条互补的寡核苷酸引物之间因部分或完全互补而形成的双链区域。该区域起始于任一引物的3'端的非互补区域,并可能延伸至另一引物的3'端或任何其他位置,终止于互补配对的结束点。在此区段内,允许存在个别碱基的错配现象,但整体上保持足够的互补性以形成稳定的双链结构。gc-count表示二聚体结合区域gc碱基个数(错配的碱基不算在内),是按其中一条引物的5’端至3’端方向第一个互补结合的碱基算作起点,最后一个互补结合的碱基算作终点,起点-终点区域可最多接受2个碱基错配;l表示二聚体结合区域从起点至终点的碱基长度;d1表示二聚体结合区域的起点至其最近一端引物的3'端的距离,d2表示二聚体结合区域的终点至其最近一端引物的3'端的距离。
46、以上指标共同参与二聚体评价,能更全面评估二聚体结合区域的难易程度、对整个体系的影响大小。最终每条候选引物都会有一个评分,即该条引物与体系中其他引物所有可能产生的二聚体的打分总和,评分越高,二聚体结合情况越严重,研究人员可直接根据评分进行筛选和组合,比常规的评价算法更灵活。
47、基于本发明提供的二聚体评价方法,当某对引物中任意一条引物的总评分大于等于103,则应考虑在体系中去除掉该对引物。例如,下文所示的f-primer和r-primer利用本发明提供的二聚体评分均为4096,应评估为需要淘汰的引物对。
48、score: 6, delta g = - 8.17 kcal/mol
49、f-primer: cgttcagtacaatgcggccg
50、r-primer:gccggcgtaacatgacttgc
51、上述引物下划线部分为二聚体接合区,结合长度为6个碱基,且都为gc碱基,二聚体结合自由能是-8.17 kcal/mol,小于本领域技术人员普遍规定的阈值-4 kcal/mol,属于在真实世界中容易结合的二聚体。可见本发明提供的二聚体评价方法可以客观评价二聚体结合的情况,并以此进行引物的筛选。
52、需要补充说明的是,本技术所要求保护的技术方案中,以引物覆盖度、特异性和二聚体结合评分的顺序作为优选评价方式,亦即,在先评价筛得的引物对用于在后评价。尽管如此,本领域技术人员在必要的情况下可随意更换上述内容的评价顺序,都在本技术的保护范围内。
53、本发明提供的引物设计方法包括确定目标病原的最佳参考基因组,搭建病原的高质量基因组库,设计引物,评价引物对的覆盖度、特异性和二聚体结合情况。该方法先对目标病原的最佳参考基因组遍历设计引物对,再将引物双端比对至基因库计算覆盖度,从而避免在获取种内基因组较多样物种特异序列时的大量重复劳动;然后检验候选引物对的特异性,确保引物对在真实样本中拷贝的片段可正确鉴定所属物种;最后再用自研的二聚体评分算法,对候选引物对进行评分,减少引物体系中的二聚体结合,提高引物体系工作效率。
本文地址:https://www.jishuxx.com/zhuanli/20250117/355917.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。