MoSi2N4掺杂过渡金属原子促进CO2还原为CO的催化剂设计方法
- 国知局
- 2024-07-12 10:39:14
本发明涉及计算材料化学,尤其涉及一种mosi2n4掺杂过渡金属原子促进co2还原为co的催化剂设计方法。
背景技术:
1、电化学二氧化碳还原反应(co2rr)是解决二氧化碳(co2)排放和利用问题的方案之一,它可以通过多个质子化步骤将co2转化为高附加值的化学燃料。然而,co2分子中的c=o键非常稳定,需要很高的能量才能断裂,这使得co2rr面临反应速率慢和法拉第效率低的问题。同时,具有多种反应产物的co2rr以及过程中不可避免存在的竞争性析氢反应(her)也导致产物选择性较低。在多种产物转化中,co作为各种化学产品和工业反应的原料具有非常高的工业价值。au和ag等贵金属催化剂是将co2选择性还原为co并有效抑制her的优良催化剂,但由于成本过高,其大规模应用受到限制。因此,开发低成本、高效稳定且不含贵金属的co2rr电催化剂仍是当前研究的重点。单原子催化剂(sac)具有一系列优异特性,如极高的原子利用效率、高产物选择性和催化活性位点分布均匀等。其中,单原子催化剂的活性与中心原子的配位环境密切相关。自石墨烯发现以来,二维(2d)材料因其特殊的结构特征和独特的物理、化学和电子特性,一直被认为非常有希望成为负载高活性过渡金属原子的sac基底,以开发出能够替代贵金属的新型co催化剂。mosi2n4是一种通过化学气相沉积法实验合成的新型二维半导体材料,由七层原子(n-si-n-mo-n-si-n)组成,具有非常高的环境稳定性。然而原始mosi2n4的co2rr活性并不好,因此便需要通过各种手段来调控其表面配位环境,激活其co2rr活性。
2、在此背景下,本发明通过在mosi2n4表面掺杂高反应活性的过渡金属原子从而设计了一系列潜在的co2rr催化剂,并根据相应步骤筛选出一种具有高稳定性、高co催化活性和选择性的单原子催化剂,有效地解决了这一问题。
技术实现思路
1、本发明在实验合成的mosi2n4的基础上,基于密度泛函理论(dft)研究了一系列用于co2rr的过渡金属原子掺杂的二维mosi2n4单原子催化剂tm@mosi2n4-nv(tm=sc-zn)。本发明首先分析了它们的稳定性。然后计算产生co的反应路径,研究了相对于hcooh、ch4和h2的产物选择性,并分析了产生co的最佳催化剂的催化性能来源。
2、为了实现上述目的,本发明提供了mosi2n4掺杂过渡金属原子促进co2还原为co的催化剂设计方法,包括以下步骤:
3、(1)构建包含活性单原子中心的催化剂计算模型
4、包括模型选定、建立单层超晶胞、设置真空层以及模型优化几个步骤。
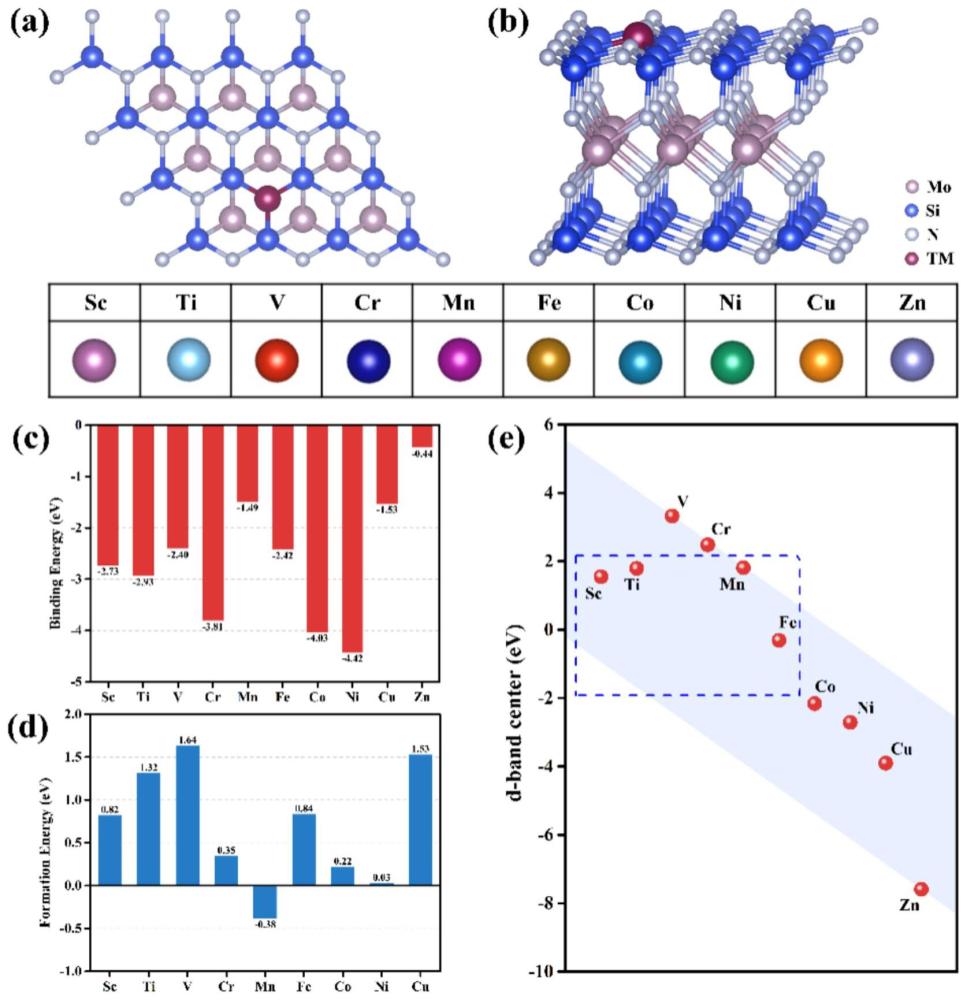
5、所述模型是由七层原子(n-si-n-mo-n-si-n)组成的二维层状结构,所述单层超晶胞为过渡金属掺杂后的3×3×1的mosi2n4超晶胞,如图1(a)和(b)所示;所述真空层为所述模型优化是指在经过固定z轴优化x轴和y轴进行的晶格参数的优化和晶胞内原子在x,y,z三个方向作用力的优化,得到tm@mosi2n4-nv单层的晶胞结构。
6、在本发明的一个实施方式中,经过模型优化后,mn@mosi2n4-nv的模型展示在图2。结构优化后mn原子并没有远离mosi2n4单层,说明mn原子与mosi2n4表面结合稳定,证明本发明的建模和优化方法是合理的。
7、(2)评估不同催化剂的热稳定性
8、热稳定性是催化剂的关键属性之一,利用公式(i)和(ii)计算得到的催化剂的结合能ebind和形成能eform来评价催化剂的热稳定性。
9、ebind=e(tm@mosi2n4-nv)-e(mosi2n4-nv)-e(tm) (i)
10、eform=e(tm@mosi2n4-nv)-e(mosi2n4-nv)-μ(tm) (ii)
11、其中e(tm@mosi2n4-nv)和e(mosi2n4-nv)分别代表tm@mosi2n4-nv单层的能量、具有n空位的mosi2n4单层的能量。e(tm)和μ(tm)分别代表真空中单个tm原子的能量和稳定体相中单个tm原子的能量。
12、催化剂的结合能ebind<0,表明催化剂结合稳定;反之,ebind>0,则催化剂结合不稳定。进一步地,通过计算催化剂的投影态密度(pdos)来分析过渡金属原子与mosi2n4表面的轨道杂化情况,验证催化剂的热稳定性。si原子的p轨道和过渡金属原子的d轨道在费米能级附近充分杂化表明了过渡金属原子与mosi2n4表面结合稳定,也证明了催化剂的热稳定性。
13、催化剂的形成能可以判断其在实验上合成的困难程度。催化剂的形成能eform<0,表明催化剂容易在温和的实验条件下能够合成。然而,一个体系的形成能为正值并不意味着该体系无法通过实验合成。例如co-mos2的形成能计算值为1.80ev,但是它能够在比较温和的水热条件下合成出来。如果催化剂的形成能都小于这个值,表明它们无需苛刻的实验条件也能合成。
14、(3)评估催化剂的co2活化能力
15、co2rr的第一步也是最关键的一步是二氧化碳分子的初始活化,即co2的吸附。而二氧化碳的吸附构型会对后续的还原过程产生很大影响,考虑两种co2初始吸附构型,分别是co2竖直初始吸附和co2水平初始吸附。通过结构优化后co2的变形程度来判断co2是否被活化,如果co2由线型变为v型(∠o-c-o<180°),则能够初步判断co2分子已被活化。同时,根据公式(iii)来计算两种co2初始吸附构型的co2吸附能来进一步判断后续研究该使用哪种构型。co2变形程度大且co2吸附能更负的吸附构型被用于后续的研究。
16、eads=e(co2_tm@mosi2n4-nv)-e(tm@mosi2n4-nv)-e(co2) (iii)
17、其中,e(co2_tm@mosi2n4-nv)是指催化剂吸附co2的体系总能量,e(tm@mosi2n4-nv)是指tm@mosi2n4-nv单层的能量,e(co2)是指单个co2分子的能量。
18、过渡金属原子的吸附能力可以用d带中心来描述。根据经典的d带中心理论,d带中心值越接近费米级,吸附能力就越强。在本发明的一个实施方式中,本发明计算了tm@mosi2n4-nv(tm=sc-zn)单层中tm原子的d带中心,发现sc,ti,mn,fe@mosi2n4-nv单层的d带中心比较接近费米能级,因此本发明预测它们的吸附能力会更强,而结合co2吸附构型的优化结果和co2吸附能计算结果也验证了本发明的预测。
19、(4)co2rr催化剂的初筛选
20、co作为各种化学产品和工业反应的原料具有非常高的工业价值。然而,以往的研究表明,当二氧化碳分子吸附在催化剂表面后,发生的co2rr可产生多种产物,包括双电子产物co和hcooh、六电子产物ch3oh、八电子产物ch4以及通过c-c偶联生成的c2+产物。本文不考虑c2+产物,因为c-c偶联需要两个或更多的活性位点,这在单原子催化剂中难以实现。如图所示,在co2rr过程中,双电子产物co和hcooh分别通过co2→*cooh→*co→co和co2→*ocho→*hcooh→hcooh路径产生。而*co和*hcooh除了解吸之外,还能够产生*cho中间物,它是产生ch4等多电子产物的关键中间物。此外,her会消耗催化剂表面的活性位点从而降低反应速率,是co2rr的主要竞争反应。因此,理想的co催化剂不仅应具有高的co2rr活性,还需要能够抑制其他副反应,如产生hcooh、*co的进一步氢化以及her。在这里,本发明首先评估tm@mosi2n4-nv单层上产生co的活性。
21、由上面的分析可知,co可按照以下步骤生成,即
22、*+co2+h++e-→*cooh (1)
23、*cooh+h++e-→*co+h2o (2)
24、*co→*+co (3)
25、为了评估催化剂产生co的催化活性,筛选优秀的co催化剂,本发明先计算了它们产生co的自由能图。根据公式(iv)计算每一步的自由能变化,建立在外加电位为0v下的co2还原为co的反应路径自由能图。
26、每个基元反应的自由能基于等人提出的计算氢电极(che)模型计算。通过自由能公式(iv)计算co2还原过程中任意两步的自由能变化δg,即
27、δg=δe+δzpe-tδs (iv)
28、其中,δe是通过dft计算得出的每个反应步骤的电子能量差值。t为温度(298.15k),δzpe和δs分别是零点能变化和熵变化,可通过频率计算获得。
29、具体包括,以*+co2、*cooh、*co、*+co为横坐标,co2还原为co的反应路径中不同步骤的自由能变化为纵坐标建立在外加电压为0v下的co2还原为co的反应路径自由能图。理论ul已被证明是用于评估催化剂活性的有效指标。因此,根据自由能图找到具有最大自由能变化值(δgmax)的步骤,然后根据公式(v)计算ul。比较tm@mosi2n4-nv单层产生co的ul,筛选出ul最接近于0的催化剂,即具有最佳的co催化活性。
30、ul=-δgmax/e (v)
31、其中,δgmax是所有基元反应中最大的自由能变化。
32、在本发明的一个实施方式中,如图6(b)所示,计算结果表明tm@mosi2n4-nv(tm=sc,ti,mn)三种催化剂产生co的ul相对较小,分别是-0.28v、-0.34v和-0.16v。这说明它们将co2还原为co所需的外加电压相对较低,反应活性更好。
33、(5)产物选择性分析:比较产生co和产生hcooh、ch4以及h2的选择性
34、作为co2rr第一步的co2分子加氢有以下两条路径:
35、co2+h++e-→*cooh和co2+h++e-→*hcoo,分别通往co和hcooh产物。因此本发明比较了催化剂产生*cooh和*hcoo的自由能变化,并且进一步计算了*hcoo产生*hcooh的自由能变化,来全面评估催化剂对于co和hcooh的产物选择性。
36、同时,确认了催化剂对于这两种产物的选择性之后,产物能否顺利脱附以及是否更倾向于进一步氢化还原仍然需要考虑。也就是说,*co形成后,除了能够发生*co解吸产生co产物,还可能进行*co继续加氢来产生c1产物的反应。而*cho是产生ch4等c1产物的关键中间物,因此本发明主要考虑*co解吸和*co加氢生成*cho中间物的反应倾向性,并且计算了步骤(4)筛选的具有更佳co催化活性的这些催化剂的co结合能和它们产生ch4的自由能图。
37、利用公式(vi)计算co结合能eb(*co):
38、eb(*co)=e(*co)-e(*)-e(co2)+e(h2o)-e(h2) (vi)
39、其中,e(*co)是吸附了*co中间体的tm@mosi2n4-nv的dft总能量,e(*)是清洁的tm@mosi2n4-nv表面的能量。e(co2)、e(h2o)和e(h2)分别是气相co2、h2o和h2的dft总能量。
40、最后,众所周知,co2rr过程中最可能发生的竞争反应是氢化反应,即*+h++e-→*h。然而,在co2rr应用中,h原子的吸收是不可避免的,但初始氢化步骤必须更有利于*cooh或*ocho的形成。为了评估催化剂的co2rr与her的反应选择性,本发明比较了co2rr的第一步加氢和her的第一步反应的自由能变化,并且结合催化剂产生co和h2的限制电位ul的差值,即ul(co2rr)-ul(h2),来更进一步评估催化剂相对于co和h2的产物选择性。
41、在经过上述筛选过程之后,本发明能够筛选出具有优秀co催化活性和产物选择性的催化剂。
42、(6)催化活性来源分析
43、本发明进一步探究了具有最佳co催化活性的催化剂的活性来源。通过比较co2吸附能与电荷转移来分析该催化剂对co2的活化能力,并且对该催化剂吸附co2后的pdos进行分析,确定co2与催化剂的相互作用。因为过渡金属原子在吸附co2后,co2的成键轨道会分裂并与过渡金属原子的3d轨道重叠,使得co2的πg轨道上的电子转移到过渡金属原子的空的d轨道上。这种电子接收、反馈和d轨道占据的协同效应确保了二氧化碳的稳定吸附和有效活化,也会造成催化活性的差异。
44、在本发明的一个实施方式中,mn@mosi2n4-nv单层具有最佳的co催化活性。如图11所示,从吸附co2的pdos上发现,在吸附co2后,co2的成键轨道会分裂并与mn原子的3d轨道重叠,使得co2的πg轨道上的电子转移到mn原子的空的d轨道上。同时,在费米能级附近存在空的反键轨道,使mn@mosi2nx-nv更容易接受电子并继续氢化,从而降低了进一步氢化的难度。
45、本发明的一种mosi2n4表面掺杂过渡金属原子来促进co2还原成co的催化剂设计方法,通过对掺杂结构进行理论建模和结构优化,设计了一系列潜在的高co催化活性的单原子催化剂。在传统密度反应理论计算的基础上,应用先进的计算方法,包括dft+u、范德华力修正对这些催化剂的热稳定性、co催化活性和产物选择性进行了合理评估。综合上述步骤的渐进式筛选,缩小研究模型范围,快速准确定位所需模型,得到理论催化性能优异的co催化剂,为实验制备高稳定性、高co催化活性和选择性的催化剂提供设计指导。
本文地址:https://www.jishuxx.com/zhuanli/20240614/88310.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表