包括将所述原料与含有有机添加剂的催化剂前体混合的重质烃原料的混合沸腾-夹带床加氢转化的制作方法
- 国知局
- 2024-07-29 09:54:52
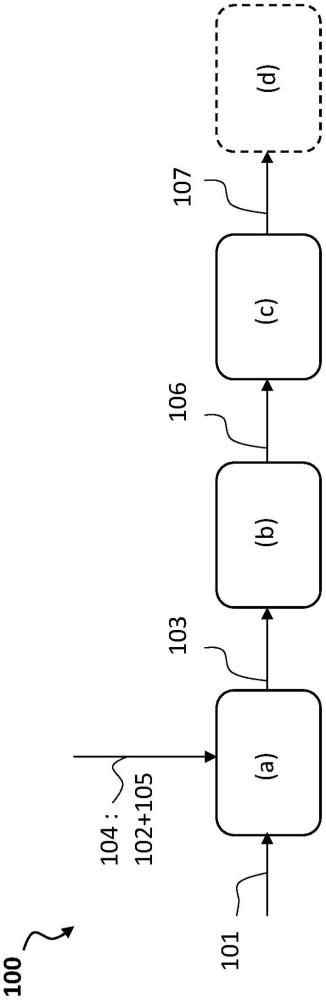
发明主题和概述在上述背景下,本发明的目的是提供一种混合加氢转化方法,其采用通过使用可溶催化前体形成的胶体或分子催化剂,解决了尤其在加氢转化反应器上游的设备中,特别是在该原料在(一个或多个)混合加氢转化反应器中转化之前的预热装置中的结垢问题。更通常地,本发明旨在提供用于重质油原料的提质的混合加氢转化方法,其实现以下中的一种或多种:更有效地处理沥青质分子,减少焦炭前体和沉积物的形成,减少设备结垢,提高转化水平,使反应器能处理更宽范围的较低品质原料,消除沸腾床反应器和下游处理设备中的无催化剂区,延长维护停机之间的操作,更有效地使用负载催化剂,提高重质油原料的吞吐量,和提高转化产物的生产速率。降低工艺容器的停机和启动的频率意味着该工艺设备的更少的压力和温度循环,并且这显著提高工艺安全性,并延长了昂贵设备的使用寿命。由此,为了实现上述目的中的至少一个,其中根据第一方面,本发明提供了一种用于重质油原料(101)的加氢转化的方法,所述重质油原料(101)含有至少50重量%的具有至少300℃的沸点的馏分,并含有金属和沥青质,所述方法包括以下步骤:(a)以使得催化剂前体配制物与硫反应时形成胶体或分子催化剂的方式将所述重质油原料与催化剂前体配制物混合来制备经调理的重质油原料,所述催化剂前体配制物包含:-包含钼的催化剂前体组合物,和-包含至少一个羧酸官能团(function)和/或至少一个酯官能团和/或酸酐官能团的有机化合物,和所述催化剂前体配制物中所述有机化合物与钼之间的摩尔比为0.1:1至20:1;(b)在至少一个预热装置中加热来自步骤(a)的所述经调理的重质油原料;(c)将来自步骤(b)的所述加热的经调理的重质油原料引入至少一个包含加氢转化多孔负载催化剂的混合沸腾-夹带床反应器中,并在氢气的存在下和在加氢转化条件下操作所述混合沸腾-夹带床反应器以产生提质的材料,并且其中该胶体或分子催化剂在步骤(b)处和/或在步骤(c)处在经调理的重质油原料中原位形成。根据一个或多个实施方案,步骤(a)包括同时将所述有机化合物与所述催化剂前体组合物(优选预先用烃油稀释剂稀释)和与所述重质油原料(优选低于大部分的催化剂前体组合物开始热分解的温度,如在室温至300℃的温度下)混合并持续1秒至30分钟的时间段。根据一个或多个实施方案,步骤(a)包括(a1)将所述有机化合物与所述催化剂前体组合物预混合以产生所述催化剂前体配制物,和(a2)将所述催化剂前体配制物与所述重质油原料混合。根据一个或多个实施方案,在步骤(a1)处,在低于大部分催化剂前体组合物开始热分解的温度的温度下、优选在室温至300℃的温度下混合所述催化剂前体组合物。根据一个或多个实施方案,烃油稀释剂用于形成催化剂前体配制物,所述烃油稀释剂优选选自减压瓦斯油、澄清油或循环油、轻质瓦斯油、减压渣油、脱沥青油和树脂。根据一个或多个实施方案,该有机化合物选自乙基己酸、环烷酸、辛酸、己二酸、庚二酸、辛二酸、壬二酸和癸二酸、辛酸乙酯、2-乙基己酸乙酯、2-乙基己酸2-乙基己酯、2-乙基己酸苄基酯、己二酸二乙酯、己二酸二甲酯、己二酸双(2-乙基己基)酯、庚二酸二甲酯、辛二酸二甲酯、辛二酸单甲酯、己酸酐、辛酸酐及其混合物。根据一个或多个实施方案,该有机化合物包含2-乙基己酸,且优选为2-乙基己酸。根据一个或多个实施方案,该有机化合物包含辛酸乙酯或2-乙基己酸2-乙基己酯,且优选为辛酸乙酯或2-乙基己酸2-乙基己酯。根据一个或多个实施方案,该催化剂前体组合物包含油溶性有机金属化合物或络合物、优选选自2-乙基己酸钼、环烷酸钼、六羰基钼,且优选为2-乙基己酸钼。根据一个或多个实施方案,所述有机化合物与所述催化剂前体配制物的钼之间的摩尔比为0.75:1至7:1、且优选1:1至5:1。根据一个或多个实施方案,胶体或分子催化剂包含二硫化钼。根据一个或多个实施方案,步骤(b)包括在280℃至450℃、更优选300℃至400℃、且最优选320℃至365℃的温度下加热。根据一个或多个实施方案,重质油原料包含重质原油、油砂沥青、常压塔底产物(bottom)、减压塔底产物、渣油、减粘裂化炉塔底产物、煤焦油、来自油页岩的重质油、液化煤、重质生物油和包含塑料废物和/或塑料热解油的重质油中的至少一种。根据一个或多个实施方案,该重质油原料具有含量大于0.5重量%的硫、至少0.5重量%的康拉逊残炭、含量大于1重量%的c7沥青质、按重量计含量大于2ppm的过渡金属和/或后过渡金属和/或准金属、和按重量计含量大于2ppm的碱金属和/或碱土金属。根据一个或多个实施方案,步骤(c)在2mpa至38mpa的绝对压力下、在300℃至550℃的温度下、在0.05h-1至10h-1的相对于每个混合反应器的体积的液体时空速lhsv下和在50至5000nm3/m3原料的与进入混合床反应器的原料混合的氢气量下进行。根据一个或多个实施方案,经调理的油原料中钼的浓度按重量计为重质油原料的5ppm至500ppm。根据一个或多个实施方案,该加氢转化多孔负载催化剂含有至少一种选自镍和钴的第viii族非贵金属、优选镍,和至少一种选自钼和钨的第vib族金属、优选钼,并包括无定形载体、优选氧化铝载体。根据一个或多个实施方案,该方法包括进一步处理提质材料的步骤(d),所述步骤(d)包括:-至少一部分或全部由加氢转化步骤(c)产生的提质材料或任选的主要在大于或等于350℃的温度下沸腾的液体重馏分在第二混合沸腾-夹带床反应器中的第二加氢转化步骤,所述液体重馏分由任选的分离步骤产生,所述任选的分离步骤分离一部分或全部由加氢转化步骤(c)产生的提质材料,所述第二混合沸腾-夹带床反应器包含第二多孔负载催化剂,并在氢气的存在下和在加氢转化条件下操作以产生具有减少的重质渣油馏分、减少的康拉逊残炭和最终减少量的硫和/或氮和/或金属的加氢转化液体流出物,-在分馏段(f)中分馏一部分或全部所述加氢转化液体流出物以产生至少一种主要在大于或等于350℃的温度下沸腾的重馏分的步骤,所述重馏分含有在大于或等于540℃的温度下沸腾的残余馏分;-用至少一种烃溶剂使一部分或全部获得的所述重馏分脱沥青以产生脱沥青油dao和残余沥青的任选步骤;以及其中,所述加氢转化步骤(c)和所述第二加氢转化步骤在2至38mpa的绝对压力下、在300℃至550℃的温度下、在0.05h-1至10h-1的相对于每个混合沸腾-夹带床反应器的体积的时空速hsv下和在50至5000nm3/m3原料的与进入每个混合沸腾-夹带床反应器的原料混合的氢气量下进行。通过阅读在本发明的具体示例性实施方案之后的描述,本发明的其它主题和优点将变得显而易见,所述实施方案通过非限制性实施例给出,该描述参考下述附图进行。附图列表图1是图示根据本发明的混合床加氢转化方法的原理的框图。图2是图示根据本发明的一个实施方案的混合床加氢转化方法的框图,其中通过将有机添加剂与催化剂前体组合物预混合来获得催化剂前体配制物。图3是图示如图2中所示的混合床加氢转化的实例的框图,其中通过将催化剂前体组合物与含有稀释剂的有机添加剂混合来获得催化剂前体配制物。图4是图示如图2中所示的混合床加氢转化的另一实例的框图,其中通过将含有催化剂前体组合物的添加剂与烃油稀释剂混合来获得催化剂前体配制物。图5是图示如图2中所示的混合床加氢转化的另一实例的框图,其中通过将稀释的催化剂前体组合物与有机添加剂混合来获得催化剂前体配制物。图6是图示根据本发明的混合床加氢转化方法和系统的实例的框图。图7是显示在根据本发明和根据现有技术的混合床加氢转化方法中制备的经调理的油原料的实例的结垢趋势的曲线图。实施方案的描述本发明的目的是提供用于改善重质油原料品质的混合床加氢转化方法和系统。用于加氢转化重质油原料的此类方法和系统使用双催化剂体系,该双催化剂体系包括分散在重质油原料中的分子或胶体催化剂,以及多孔负载催化剂。它们还使用在催化剂前体配制物中加入的有机添加剂,在一个或多个沸腾床反应器中操作该双催化剂体系之前,将该催化剂前体配制物与重质油原料混合,每个沸腾床反应器包括包含多孔负载催化剂的膨胀床的固相、包含重质油原料、分散在其中的胶体或分子催化剂和有机添加剂的液体烃相、和包含氢气的气相。本发明的混合床加氢转化方法和系统减少设备结垢,且尤其是在(一个或多个)混合加氢转化反应器上游的设备中的结垢,特别是在原料在(一个或多个)混合加氢转化反应器中的转化之前的预热设备中的结垢,并可以有效处理沥青质、减少或消除焦炭前体和沉积物的形成,尤其是通过允许加氢转化在高温下操作来提高转化水平,并消除原本将存在于(一个或多个)常规沸腾床加氢转化反应器和下游处理设备中的无催化剂区。本发明的混合床加氢转化方法和系统还允许更有效地使用多孔负载催化剂和组合的双催化剂体系。术语下文给出了一些定义,尽管在说明书中将进一步给出下文定义的主题的更多细节。术语“加氢转化”应当是指其主要目的是降低重质油原料的沸程且其中大部分原料被转化为沸程低于原始原料的产品的方法。加氢转化通常包括将较大的烃分子断裂成具有较少碳原子数和较高氢/碳比的较小分子片段。在加氢转化过程中实施的反应允许在氢的存在下主要通过碳-碳键的断裂来降低烃分子的尺寸,以饱和切断的键和芳环。发生加氢转化的机理通常包括在主要通过热裂化的断裂过程中形成烃自由基,随后在活性催化剂位点的存在下用氢将自由基末端或部分封端。当然,在加氢转化过程中,可能发生通常与“加氢处理”相关的其它反应,如从原料中除去硫和氮以及烯烃饱和。根据英语术语,术语“加氢裂化”通常用作“加氢转化”的同义词,尽管“加氢裂化”更应当是指类似于加氢转化但其中烃分子的裂化主要为催化裂化(即在具有负责裂化活性的相,例如包含在粘土或沸石中的酸性位点的加氢裂化催化剂的存在下发生的裂化)的方法。例如,根据法国术语,可以翻译为“hydrocraquage”的加氢裂化通常是指最后的定义(催化裂化),并且其使用例如只针对减压馏分油作为待转化的油原料的情况,而法国术语“加氢转化”通常只针对重质油原料如常压和减压渣油的转化(但不仅如此)。术语“加氢处理”应当是指更温和的操作,其主要目的是从原料中除去杂质如硫、氮、氧、卤化物和痕量金属,并且通过使它们与氢反应而不是令它们自身反应来饱和烯烃和/或稳定烃自由基。主要目的不是改变原料的沸程。加氢处理最通常使用固定床反应器进行,尽管其它加氢处理反应器也可用于加氢处理,其实例是沸腾床加氢处理反应器。术语“加氢处理”应广义地指“加氢转化”/“加氢裂化”和“加氢处理”方法。术语“加氢转化反应器”应指其中原料的加氢转化是主要目的的任何容器,例如在氢气和加氢转化催化剂的存在下进料的裂化(即降低沸程)。加氢转化反应器通常包括可向其中引入重质油原料和氢气的输入口,可从中取出提质材料的输出口。具体而言,加氢转化反应器的特征还在于具有足够的热能以便通过热分解将较大的烃分子断裂成较小的分子。加氢转化反应器的实例包括但不限于浆料床反应器,也称为夹带床反应器(三相——液体、气体、固体——反应器,其中固相和液相的行为可类似于均相)、沸腾床反应器(三相流化反应器)、移动床反应器(三相反应器,其固体催化剂向下运动,且液体和气体向上或向下流动)和固定床反应器(三相反应器,其中液体进料在固体负载催化剂的固定床上向下滴流,氢气通常与液体并流流动,但在一些情况下可能逆流流动)。用于加氢转化反应器的术语“混合床”和“混合沸腾床”和“混合夹带-沸腾床”应当是指除了保持在沸腾床反应器中的多孔负载催化剂之外还包含夹带催化剂的沸腾床加氢转化反应器。类似地,对于加氢转化方法,这些术语由此应当是指包括在至少同一加氢转化反应器中的沸腾床与夹带床的混合操作的方法。该混合床是具有必然不同的粒度和/或密度的两种类型的催化剂的混合床,一种类型的催化剂——“多孔负载催化剂”——保持在反应器中,而另一种类型的催化剂——“夹带催化剂”,通常也称为“浆料催化剂”——与流出物(提质的原料)一起夹带出反应器。在本发明中,夹带催化剂是下文定义的胶体催化剂或分子催化剂。术语“胶体催化剂”和“胶体分散催化剂”应当是指具有例如直径小于约100nm、优选直径小于约10nm、更优选直径小于约5nm、和最优选直径小于约1nm的粒度的催化剂颗粒(即胶体尺寸)。术语“胶体催化剂”包括但不限于分子或分子分散的催化剂化合物。术语“分子催化剂”和“分子分散的催化剂”应当是指基本上“溶解”或完全与重质油烃原料、非挥发性液体馏分、底产物馏分、渣油、或可能存在催化剂的其它原料或产物中的其它催化剂化合物或分子离解的催化剂化合物。其还应指仅含有接合在一起的少量催化剂分子(例如15个分子或更少)的非常小的催化剂颗粒或片。术语“多孔负载催化剂”、“固体负载催化剂”和“负载催化剂”应当是指通常用于常规沸腾床和固定床加氢转化系统的催化剂,包括主要设计用于加氢裂化或加氢脱金属的催化剂和主要设计用于加氢处理的催化剂。此类催化剂通常包含(i)具有大表面积和大量互连通道或孔隙的催化剂载体和(ii)分散在该孔隙内的活性催化剂如钴、镍、钨和钼的硫化物的微细颗粒。负载催化剂通常以圆柱形粒料或球形固体形式生产,尽管其它形状也是可行的。当用于描述正在施以或已经施以加氢转化的原料或所得材料或产物时,术语“提质”、“提质处理”和“提质的”应当是指以下中的一种或多种:原料分子量的降低,原料沸点范围的降低,沥青质浓度的降低,烃自由基浓度的降低,康拉逊残炭的降低,原料h/c原子比的增加,和杂质如硫、氮、氧、卤化物和金属的量的降低。术语“经调理的原料”和“经调理的重质油原料”应当是指要在至少一个加氢转化混合床反应器中处理的重质油原料,其中包含催化剂前体组合物和有机添加剂的催化剂前体配制物已充分组合和混合以便在形成催化剂时、尤其是通过与硫反应形成催化剂时该催化剂将包含分散在该原料内的胶体或分子催化剂的原料。术语“主动混合装置”应当是指包括活动部件,例如搅拌杆或推进器或涡轮推进器以主动混合各组分的混合装置。在下文中,术语“包含”与“包括”和“含有”同义(与之具有相同意思),并且是包含性的或开放性的,且不排除其它未指定的要素。应当理解的是,术语“包含”包括排他性和封闭性术语“由……组成”。术语“在……和……之间”和“在……至……的范围内”和“……至……”意味着除非另行说明,否则区间边界处的值包括在所述值的范围内。在下面的详述中,阐述了许多具体细节以传达对根据本发明的方法和系统的更深理解。但是,对本领域技术人员将显而易见的是,可以在没有所有这些具体细节的情况下使用该方法和系统。在另一些情况下,并未详述公知的特征,以免不必要地使描述复杂化。图1是示意性图示根据本发明的混合床加氢转化方法100的原理的框图。其与例如us2005/0241991中公开的常规混合床方法的不同之处特别在于,当与油原料混合时,该催化剂前体配制物包含有机添加剂,所述催化剂前体配制物还包含催化剂前体组合物,所述催化剂前体组合物包含钼且具有特定的有机添加剂与钼的摩尔比。术语“有机化合物”和“有机添加剂”在本说明书中无差别地用于表示在步骤(a)处与重质油原料混合的催化剂前体配制物中添加的包含至少一个羧酸官能团和/或至少一个酯官能团和/或酸酐官能团的有机化合物,并且在下文进一步详细描述。有机添加剂是除催化剂前体组合物中初始存在的任何可能的有机化合物之外的化合物。根据本发明,在加氢转化方法100中处理重质油原料101,所述重质油原料101含有至少50重量%的沸点为至少300℃的馏分,并含有金属和沥青质,所述方法包括以下步骤:(a)以使得催化剂前体配制物104与硫反应时形成胶体或分子催化剂的方式将所述重质油原料101与催化剂前体配制物104混合来制备经调理的重质油原料103,所述催化剂前体配制物104包含:-包含钼的催化剂前体组合物105,和-包含至少一个羧酸官能团和/或至少一个酯官能团和/或酸酐官能团的有机化合物102,和所述有机化合物102与钼之间的摩尔比为0.1:1至20:1;(b)通过至少一个预热装置加热来自步骤(a)的经调理的重质油原料103;(c)将来自步骤(b)的加热的经调理的重质油原料106引入至少一个包含加氢转化多孔负载催化剂的混合沸腾-夹带床反应器中,并在氢气的存在下和在加氢转化条件下操作所述混合沸腾-夹带床反应器以产生提质的材料107。该提质的材料107可以在任选的步骤(d)中进一步处理。在根据本发明的加氢转化方法中,在步骤(b)和/或步骤(c)处在经调理的重质油原料中原位形成胶体或分子催化剂。现在在下面详细描述涉及的每个步骤、料流和材料。下面提到的一些参考数字涉及图6,其示意性图示了根据本发明的混合床加氢转化系统600的实例,所述系统在一般方法的描述之后在说明书下文中详细描述。重质油原料术语“重质油原料”应指重质原油、油砂沥青、精炼过程遗留的桶底物和渣油(例如减粘裂化炉底产物)和任何其它低品质材料,其含有大量的高沸点烃馏分和/或包括大量的沥青质,其可使固体负载催化剂失活和/或引起或导致焦炭前体和沉积物的形成。重质油原料101由此可包含至少一种以下原料:重质原油、油砂沥青、常压塔底产物、减压塔底产物、渣油、减粘裂化炉塔底产物、煤焦油、来自油页岩的重质油、液化煤、重质生物油和包含塑料废物和/或塑料热解油的重质油。塑料热解油是由塑料、优选塑料废物的热解获得的油,并且可以获自热、催化热解处理,或者可以通过加氢热解(在催化剂和氢气的存在下的热解)来制备。特别地,经处理的重质油原料含有烃馏分,其至少50重量%、优选至少80重量%具有至少300℃、优选至少350℃或至少375℃的沸点。这些是原油或来自原油的常压和/或减压蒸馏的重质烃馏分。它们也可以是常压和/或减压渣油,且特别是来自加氢处理、加氢裂化和/或加氢转化的常压和/或减压渣油。其还可以是真空馏分油、来自催化裂化单元如流化催化裂化(fcc)、焦化或减粘裂化单元的馏分。优选地,它们是减压渣油。通常,这些渣油是其中至少80重量%具有至少450℃或更高、且最通常至少500℃或540℃的沸点的馏分。从润滑油生产单元提取的芳族馏分、脱沥青油(来自脱沥青单元的残油)和沥青(来自脱沥青单元的渣油)也适合作为原料。该原料还可以是来自直接煤液化的残余馏分(来自例如注册商标h-coal工艺的减压馏分油和/或常压和/或减压渣油)、煤热解或页岩油渣油、或来自单独或与煤和/或石油馏分混合的木质纤维素生物质的直接液化的残余馏分(本文中称为“重质生物油”)。重质油原料的实例包括但不限于lloydminster重质油、冷湖沥青、athabasca沥青、urals原油、阿拉伯重质原油、阿拉伯轻质原油、常压塔底产物、减压塔底产物、渣油(或“resid”)、渣油沥青、减压渣油、溶剂脱沥青沥青(solvent deasphalting pitch)和在对原油、来自焦油砂的沥青、液化煤、油页岩或煤焦油原料施以蒸馏、热分离等之后残留的且含有较高沸点馏分和/或沥青质的非挥发性液体馏分。所有这些原料可以单独使用或混合使用。在根据本发明的方法和系统中处理的上述重质油原料含有金属和沥青质,特别是c7沥青质,以及其它杂质如硫和氮。术语“沥青质”应当是指重质油原料的馏分,其通常不溶于石蜡族溶剂如丙烷、丁烷、戊烷、己烷和庚烷,并且包括通过杂原子如硫、氮、氧和金属保持在一起的缩合环状化合物的板片。沥青质广泛地包括多种多样的具有80至160,000个碳原子的复杂化合物。沥青质在操作上被限定为“c7沥青质”,即根据标准astm d 6560(也对应于标准nf t60-115)的庚烷不溶性化合物,并且在本说明书中关于沥青质的任何内容是指c7沥青质。c7沥青质是已知通过它们形成重质烃残余物(通常称为焦炭)的能力和通过它们产生严重限制加氢处理和加氢转化单元可操作性的沉积物的倾向来抑制残余馏分的转化的化合物。重质油原料101通常可具有含量大于0.5重量%的硫、至少3重量%的康拉逊残炭、含量大于1重量%的c7沥青质、含量按重量计大于2ppm的过渡金属和/或后过渡金属和/或准金属、以及含量按重量计大于2ppm的碱金属和/或碱土金属。这些类型的原料实际上通常富含杂质,如金属,特别是过渡金属(例如ni、v)和/或后过渡金属,和/或准金属,其含量按重量计可以大于2ppm、或按重量计大于20ppm、且按重量计甚至大于100ppm,以及碱金属(例如na)和/或碱土金属,其含量按重量计可以高于2ppm、按重量计甚至高于5ppm、且按重量计甚至高于7ppm或10ppm。硫含量实际上通常高于0.5重量%、且甚至高于1重量%、或甚至大于2重量%。c7沥青质的含量实际上可以为至少1重量%、且甚至高于3重量%。康拉逊残炭实际上通常高于3重量%、且甚至至少5重量%。康拉逊残炭由astm d482标准定义,且表示在标准的温度和压力条件下热解后产生的残炭的量。这些含量以进料总重量的重量%表示。步骤(a):经调理的重质油原料制备步骤(a)包括以使得催化剂前体配制物104与硫反应时将形成胶体或分子催化剂的方式将所述重质油原料101与催化剂前体配制物104混合。这种共混形成了本文中称为经调理的重质油原料103的物质。催化剂前体配制物104包含含有钼的催化剂前体组合物105和含有至少一个羧酸官能团和/或至少一个酯官能团和/或酸酐官能团的有机化合物102。所述有机化合物102和钼之间的摩尔比为0.1:1至20:1。该步骤包括与催化剂前体配制物的彻底混合,这将导致形成分散在重质油中的胶体或分子催化剂。根据一个或多个实施方案,烃油稀释剂用于形成催化剂前体配制物104。优选地,所述烃油稀释剂选自减压瓦斯油、澄清油或循环油、轻质瓦斯油、减压渣油、脱沥青油和树脂,如下文进一步详述的那样,并且优选为减压瓦斯油。发明人已经表明,该混合步骤(a)改进了混合沸腾-夹带床加氢转化方法,特别是通过减少设备的结垢,尤其是在混合加氢转化反应器上游在步骤(b)的原料加热设备中的结垢。不受任何理论的束缚,在混合重质油原料与催化剂前体组合物的过程中有机添加剂的存在使得胶体或分子催化剂前体更好地溶解在进料中,避免或减少混合加氢转化反应器上游的设备中,如加热设备中特别由金属沉积造成的结垢,并因此改善在步骤(b)处和/或在后面的阶段形成的胶体或分子催化剂的分散,由此产生金属活性位点的更大可用性,有利于作为焦炭和沉积物的前体的自由基的氢化,并显著减少设备的结垢。有机添加剂具有至少一个羧酸官能团和/或至少一个酯官能团和/或酸酐官能团的有机添加剂102优选包含至少6个碳原子、且更优选至少8个碳原子。通常,有机添加剂102既不是催化剂前体也不是催化剂。特别地,有机添加剂102不含任何金属。有机添加剂的实例包括但不限于2-乙基己酸、环烷酸、辛酸、己二酸、庚二酸、辛二酸、壬二酸、癸二酸、辛酸乙酯、2-乙基己酸乙酯、2-乙基己酸2-乙基己酯、2-乙基己酸苄酯、己二酸二乙酯、己二酸二甲酯、己二酸双(2-乙基己基)酯、庚二酸二甲酯、辛二酸二甲酯、辛二酸单甲酯、己酸酐、辛酸酐。有利地,该有机添加剂是选自上文列举的具体化合物列表及其混合物中的有机化合物。优选地,有机添加剂是包含至少一个羧酸官能团的有机化合物、且更优选选自2-乙基己酸、环烷酸、辛酸、己二酸、庚二酸、辛二酸、壬二酸和癸二酸。更优选地,有机添加剂包含2-乙基己酸或由2-乙基己酸组成。有机添加剂可以是包含至少一个酯官能团和/或酸酐官能团的有机化合物,并例如选自辛酸乙酯、2-乙基己酸乙酯、2-乙基己酸2-乙基己酯、2-乙基己酸苄酯、己二酸二乙酯、己二酸二甲酯、己二酸双(2-乙基己基)酯、庚二酸二甲酯、辛二酸二甲酯、辛二酸单甲酯,和/或选自己酸酐和辛酸酐。更优选地,包含至少一个酯官能团和/或酸酐官能团的有机添加剂包含或由辛酸乙酯或2-乙基己酸2-乙基己酯或其混合物组成,并优选为辛酸乙酯或2-乙基己酸2-乙基己酯。加入有机添加剂,使得催化剂前体配制物104中有机添加剂对钼(由催化剂前体化合物,例如2-乙基己酸钼产生)的摩尔比为约0.1:1至约20:1、优选约0.75:1至约7:1、且更优选约1:1至约5:1。术语“约”应当是指±5%、优选±1%的近似值。催化剂前体配制物催化剂前体配制物包含催化剂前体组合物,其选自本领域技术人员已知的所有含钼的金属催化剂前体,能够在氢和/或h2s和/或任何其它硫源的存在下形成胶体或分子分散的催化剂(即浆料催化剂),并能够在注入所述重质油原料后将重质油原料加氢转化。含钼的催化剂前体组合物有利地是至少含有一种过渡金属的油溶性催化剂前体组合物。催化剂前体组合物优选包含油溶性有机金属化合物或络合物。油溶性催化剂前体组合物优选具有100℃至350℃、更优选150℃至300℃、且最优选175℃至250℃的分解温度(该催化剂前体组合物基本上化学稳定的温度)。油溶性有机金属化合物或络合物优选选自2-乙基己酸钼、环烷酸钼和六羰基钼。这些化合物是油溶性催化剂前体组合物的非限制性实例。目前优选的催化剂前体组合物是2-乙基己酸钼(通常也称为辛酸钼)。通常,2-乙基己酸钼含有15重量%的钼,并且具有足够高的分解温度或范围,以避免在低于250℃的温度下与重质油原料混合时的显著热分解。本领域技术人员可以遵循本公开选择混合温度曲线,由此致使所选前体组合物的混合而在形成胶体或分子催化剂之前没有显著的热分解。并入有机添加剂混合步骤(a)可以根据下文详细描述的不同方式进行,主要取决于有机添加剂是与重质油原料和催化剂前体组合物同时混合,还是以顺序方式引入,特别是通过将其与重质油原料混合之前将催化剂前体组合物与有机添加剂预混合以形成催化剂前体配制物。混合步骤(a)有利地包括操作至少一个调理混合器610,该调理混合器610被配置为在原料与催化剂前体配制物104之间提供彻底混合以形成经调理的重质油原料。第一实施方案:同时混合油原料、有机添加剂和催化剂前体组合物根据第一实施方案,步骤(a)包括同时将有机添加剂102与催化剂前体组合物105(优选预先用烃油稀释剂稀释)和与重质油原料101混合。根据该实施方案,在与重质油原料101混合过程中,由此形成包含催化剂前体组合物105(优选预先稀释)和有机添加剂102的催化剂前体配制物104。如前所述,加入有机添加剂,使得有机添加剂对钼(由催化剂前体组合物,例如2-乙基己酸钼引入)的摩尔比为约0.1:1至约20:1、优选约0.75:1至约7:1、且更优选约1:1至约5:1。此类同时混合优选在低于大部分催化剂前体组合物开始热分解的温度下进行,如在室温(例如15℃)至300℃、更优选50℃至200℃、且甚至更优选75℃至175℃的温度下进行。此类同时混合进行足够长的时间,并以使催化剂前体甚至更优选遍布该原料分散的方式进行,以便获得经调理的重质油原料103,其中催化剂前体组合物在重质油原料中充分混合。优选地,表压为0mpa至25mpa、更优选0.01mpa至5mpa。为了在形成胶体或分子催化剂之前获得催化剂前体组合物在重质油原料中的充分混合,重质油原料101、有机添加剂102和有利地用烃稀释剂稀释的催化剂前体组合物105的同时混合优选进行1秒至30分钟、更优选1秒至10分钟、且最优选2秒至3分钟的时间段。在本说明书中,1秒的混合时间(或用于混合的停留时间)包括瞬时混合。尽管将催化剂前体组合物105与重质油原料101和有机添加剂102直接共混也在本发明的范围内,但在此类情况下必须小心混合这些组分足够长的时间,以便在形成催化剂之前在原料中彻底共混该催化剂前体组合物。但是,长时间混合,例如24小时混合,可能使得某些工业操作过于昂贵。因此,根据第一实施方案,步骤(a)优选包括在与重质油原料101和有机添加剂102同时混合之前稀释催化剂前体组合物105:用烃稀释剂预稀释催化剂前体组合物105,随后将所述稀释的催化剂前体组合物与重质油原料和有机添加剂102同时混合,这极大地有助于在原料中彻底和精细地共混该催化剂前体组合物,特别是对于大规模工业操作要在经济上可行所需的相对短的时间段内。催化剂前体组合物、优选油溶性催化剂前体组合物与稀释剂烃料流的此类混合例如描述在us2005/0241991中并在下文中回顾。提供稀释的催化剂前体组合物通过以下来缩短总混合时间:(1)减少或消除极性更大的催化剂前体组合物与重质油原料之间溶解度的差异,(2)减少或消除催化剂前体组合物与重质油原料之间的流变学差异,和/或(3)打断催化剂前体分子以形成烃油稀释剂中的溶质以更容易分散在重质油原料中。在其中重质油原料含有水(例如冷凝水)的情况下,首先形成稀释的催化剂前体组合物是特别有利的。否则,水对于极性催化剂前体组合物的更大亲和力可能引起催化剂前体组合物的局部附聚,导致差的分散和形成微米尺寸或更大的催化剂颗粒。烃油稀释剂优选基本不含水(即含有小于0.5重量%的水、优选小于0.1重量%的水、且更优选按重量计小于750ppm的水),以防止形成大量的微米级或更大的催化剂颗粒。合适的烃稀释剂的实例包括但不限于称为“vgo”的减压瓦斯油(其通常具有360℃-524℃的沸程)、澄清油或循环油(其通常具有360℃-550℃的沸程)、轻质瓦斯油(其通常具有200℃-360℃的沸程)、减压渣油(其通常具有524℃+的沸点)、脱沥青油和树脂。催化剂前体组合物105对烃油稀释剂的质量比优选为1:500至1:1、更优选1:150至1:2、且最优选1:100至1:5(例如1:100、1:50、1:30、或1:10)。在同时混合之前,所述稀释有利地进行1秒至30分钟、优选1秒至10分钟、且最优选2秒至3分钟的的时间段。该稀释的实际时间至少部分取决于温度(也就是说,其影响流体的粘度)和为了稀释而进行的混合的强度。所述稀释还有利地在低于大部分催化剂前体组合物开始热分解的温度下进行,优选在室温(例如15℃)至300℃、更优选室温至200℃、甚至更优选50℃至200℃、最优选75℃至150℃、且甚至更优选75℃至100℃的温度下进行。应当了解,形成稀释的催化剂前体组合物105的实际温度通常主要取决于所用的特定前体组合物的分解温度。调理混合器610可包括如下文详述的主动混合装置、用于管道的任何注入系统或任何在线混合器。根据第一实施方案的步骤(a)的同时混合可以在构成调理混合器610的主动混合装置的专用容器中进行。此类配置特别可以提高在随后阶段中形成的胶体或分子催化剂的分散。使用专用容器也能够实现长的停留时间。此类同时混合可以替代性地包括将所述有机添加剂102和优选预先用烃油稀释剂稀释的催化剂前体组合物105注入向混合沸腾-夹带床反应器输送重质油原料101的管道中。该调理混合器610由此在此类配置中包括在其中进行混合的(一个或多个)管道部分,并可能包括附加系统以帮助混合,例如进一步描述的静态在线混合器或高剪切在线混合器。与在专用容器中混合相比,此类配置可特别减少设备投资和所需占地面积。用于同时混合的调理混合器610也可包括主动混合装置的此类专用容器和可能包括静态和/或高剪切在线混合器的管内注入系统的组合。可用于实现催化剂前体组合物105(优选稀释的)与重质油原料101和有机添加剂102的彻底同时混合的混合设备的实例包括但不限于高剪切混合,如在具有螺旋桨或涡轮叶轮的泵中产生的混合;多个静态在线混合器;与在线高剪切混合器组合的多个静态在线混合器;与在线高剪切混合器组合的多个静态在线混合器;与在线高剪切混合器和随后在缓冲容器中周围的泵组合的多个静态在线混合器;上述和随后的一个或多个多级离心泵的组合。根据一个实施方案,可以使用具有多个腔室的高能泵进行连续混合而非间歇式混合,作为泵送过程本身的一部分,在所述多个腔室中搅拌和混合催化剂前体组合物105(优选稀释的)、重质油原料101与有机添加剂102。上述混合设备也可用于上述稀释阶段,其中催化剂前体组合物105与烃油稀释剂混合。增加同时混合过程的活力和/或剪切能通常减少实现彻底混合所需的时间。第二实施方案:预混合催化剂前体组合物与有机添加剂根据第二实施方案,如图2中示意性所示,混合步骤(a)包括(a1)将有机添加剂化合物102与催化剂前体组合物105预混以产生催化剂前体配制物104,和(a2)将所述催化剂前体配制物104与所述重质油原料101混合。预混合有机添加剂化合物102与催化剂前体组合物105以产生催化剂前体配制物104的步骤(a1)可异位(即在加氢转化系统外部)进行。在此类第二实施方案中,调理混合器610包括用于步骤(a1)的至少第一混合设备和用于步骤(a2)的至少第二混合设备。在步骤(a1)处,加入有机添加剂,使得催化剂前体配制物104中有机添加剂102对钼(由催化剂前体组合物,例如2-乙基己酸钼引入)的摩尔比为约0.1:1至约20:1、优选约0.75:1至约7:1、且更优选约1:1至约5:1。在步骤(a1)处,在低于大部分催化剂前体组合物开始热分解的温度下,优选在室温(例如15℃)至300℃、优选在室温至200℃、甚至更优选50℃至200℃、更优选75℃至150℃、且甚至最优选75℃至100℃的温度下混合催化剂前体组合物105。步骤(a1)步骤(a1)本身可以以下面详述的不同方式进行。尽管在步骤(a2)中将由催化剂前体组合物105和有机添加剂102组成的催化剂前体配制物与重质油原料101直接共混在本发明的范围内,但根据本发明的所述第二实施方案的方法优选包括:在步骤(a1)处使用烃油稀释剂来产生催化剂前体配制物104,尤其有助于在步骤(a2)处在对于大规模工业操作要在经济上可行所需的相对短的时间段内在原料中彻底和精细地共混该催化剂前体组合物。出于上文就第一实施方案的稀释催化剂前体组合物的描述已给出的原因(减少或消除溶解度、流变学等的差异),使用烃油稀释剂来产生催化剂前体配制物104缩短了步骤(a2)处的混合时间。合适的烃稀释剂的实例包括但不限于称为“vgo”的减压瓦斯油(其通常具有360℃-524℃的沸程)、澄清油或循环油(其通常具有360℃-550℃的沸程)、和轻质瓦斯油(其通常具有200℃-360℃的沸程)。催化剂前体配制物104中催化剂前体组合物105对烃油稀释剂的质量比优选为1:500至1:1、更优选1:150至1:2、且最优选1:100至1:5(例如1:100、1:50、1:30、或1:10)。根据一个或多个子实施方案,如图3中示意性所示,根据所述第二实施方案的方法300的步骤(a1)包括:-(α1)将所述有机添加剂102与烃油稀释剂108预混合以形成含有添加剂的稀释剂108’;和-(α2)将所述含有添加剂的稀释剂108’与所述催化剂前体组合物105混合以产生所述催化剂前体配制物104。步骤(α1)优选在室温(例如15℃)至300℃、优选室温至200℃、甚至更优选50℃至200℃、最优选75℃至150℃、且甚至最优选75℃至100℃的温度下进行。预混合阶段(α1)的压力也有利地为稀释剂料流108的实际压力。优选地,预混合阶段(α1)的表压为0mpa至25mpa、更优选0.01mpa至5mpa。停留时间可以为1秒至几天、优选1秒至30分钟、更优选1秒至10分钟、且最优选1秒至30秒。步骤(α2)优选在低于大部分催化剂前体组合物105开始热分解的温度下进行,优选在室温(例如15℃)至300℃、优选室温至200℃、甚至更优选50℃至200℃、最优选75℃至150℃、且甚至最优选75℃至100℃的温度下进行。混合阶段(α2)的压力也有利地为料流108’的实际压力。优选地,混合阶段(α2)的表压为0mpa至25mpa、更优选0.01mpa至5mpa。停留时间可以为1秒至几天、优选1秒至30分钟、更优选1秒至10分钟、且最优选1秒至30秒。应当了解,在步骤(α2)处操作的实际温度通常很大程度上取决于所用的特定前体组合物的分解温度。根据一个或多个子实施方案,如图4处示意性所示,根据所述第二实施方案的方法400的步骤(a1)包括:-(β1)将所述有机添加剂102与所述催化剂前体组合物105预混合以形成含有添加剂的催化剂前体组合物105’;和-(β2)将所述含有添加剂的催化剂前体组合物105’与烃油稀释剂108混合以制造所述催化剂前体配制物104。步骤(β1)优选在低于大部分催化剂前体组合物105开始热分解的温度下进行,优选在室温(例如15℃)至300℃、优选室温至200℃、甚至更优选50℃至200℃、最优选75℃至150℃、且甚至最优选75℃至100℃的温度下进行。优选地,混合阶段(β1)的表压为0mpa至25mpa、更优选0.01mpa至5mpa。停留时间可以为1秒至几天、优选1秒至30分钟、更优选1秒至10分钟、且最优选1秒至30秒。步骤(β2)优选在低于大部分催化剂前体组合物105开始热分解的温度下进行,优选在室温(例如15℃)至300℃、优选室温至200℃、甚至更优选50℃至200℃、最优选75℃至150℃、且甚至最优选75℃至100℃的温度下进行。优选地,混合阶段(β2)的表压为0mpa至25mpa、更优选0.01mpa至5mpa。停留时间可以为1秒至几天、优选1秒至30分钟、更优选1秒至10分钟、且最优选1秒至30秒。应当了解,在步骤(β1)和(β2)处所用的实际温度通常很大程度上取决于所用的特定前体组合物的分解温度。根据一个或多个子实施方案,如图5处示意性所示,根据所述第二实施方案的方法500的步骤(a1)包括:-(γ1)将所述催化剂前体组合物105与烃油稀释剂108预混合以形成稀释的催化剂前体组合物109;和-(γ2)将所述稀释的催化剂前体组合物109与有机添加剂102混合以产生所述催化剂前体配制物104。步骤(γ1)优选在低于大部分催化剂前体组合物105开始热分解的温度下进行,优选在室温(例如15℃)至300℃、优选室温至200℃、甚至更优选50℃至200℃、最优选75℃至150℃、且甚至最优选75℃至100℃的温度下进行。优选地,混合阶段(γ1)的表压为0mpa至25mpa、更优选0.01mpa至5mpa。停留时间可以为1秒至几天、优选1秒至30分钟、更优选1秒至10分钟、且最优选1秒至30秒。步骤(γ2)优选在低于大部分催化剂前体组合物105开始热分解的温度下进行,优选在室温(例如15℃)至300℃、优选室温至200℃、甚至更优选50℃至200℃、最优选75℃至150℃、且甚至最优选75℃至100℃的温度下进行。优选地,混合阶段(γ2)的表压为0mpa至25mpa、更优选0.01mpa至5mpa。停留时间可以为1秒至几天、优选1秒至30分钟、更优选1秒至10分钟、且最优选1秒至30秒。应当了解,在步骤(γ1)和(γ2)处所用的实际温度通常很大程度上取决于所用的特定前体组合物的分解温度。步骤(a1)的不同混合子步骤可以使用不同的混合设备进行,其实例包括但不限于高剪切混合,如在具有螺旋桨或涡轮叶轮的容器中产生的混合;多个静态在线混合器;与在线高剪切混合器组合的多个静态在线混合器;与在线高剪切混合器组合的多个静态在线混合器;与在线高剪切混合器和随后在缓冲容器中周围的泵组合的多个静态在线混合器;上述和随后的一个或多个多级离心泵的组合;以及一个或多个多级离心泵。根据一个实施方案,可以使用具有多个腔室的高能泵进行连续混合而非间歇式混合,作为泵送过程本身的一部分,在所述多个腔室中搅拌和混合待混合组分。例如,步骤(a1)的不同混合子步骤中的每一个可以在主动混合装置的专用容器中进行,所述主动混合装置是调理混合器610的第一混合设备的一部分。此类配置特别可以提高在随后阶段中形成的胶体或分子催化剂的分散。使用专用容器也能够实现长的停留时间。根据另一实例,步骤(a1)的不同混合子步骤中的每一个可以替代性地包括将待混合组分注入输送另一组分的管道中,在本文中称为管内注入系统。该调理混合器610的第二混合设备由此在此类配置中包括在其中进行混合的(一个或多个)管道部分,并可能包括附加系统以帮助混合,例如如上所述的静态在线混合器或高剪切在线混合器。与在专用容器中混合相比,此类配置可特别减少设备投资和所需占地面积。根据另一实例,调理混合器610的第一混合设备可包括主动混合装置的此类专用容器和可能包括静态和/或高剪切在线混合器的管内注入系统的组合。步骤(a2)将已经含有有机添加剂的催化剂前体配制物104与所述重质油原料101混合的步骤(a2)优选在低于大部分催化剂前体组合物开始热分解的温度下进行,如在室温(例如15℃)至300℃、优选50℃至200℃、且更优选75℃至175℃的温度下进行,以产生经调理的重质油原料103。优选地,表压为0mpa至25mpa、更优选0.01mpa至5mpa。步骤(a2)进行足够的时间,并以使催化剂前体配制物遍布该原料分散的方式进行,以便获得经调理的重质油原料103,其中催化剂前体组合物在重质油原料中充分混合。为了在形成胶体或分子催化剂之前获得催化剂前体配制物104在重质油原料中的充分混合,步骤(a2)优选进行1秒至30分钟、更优选1秒至10分钟、且最优选2秒至3分钟的时间。根据第二实施例的步骤(a2)可以在构成调理混合器610的第二混合设备的主动混合装置的专用容器中进行。此类配置特别可以提高在随后阶段中形成的胶体或分子催化剂的分散。使用专用容器也能够实现长的停留时间。步骤(a2)可以替代性地包括将所述催化剂前体配制物104注入向混合沸腾-夹带床反应器输送重质油原料101的管道中。该调理混合器610的第二混合设备由此在此类配置中包括在其中进行混合的(一个或多个)管道部分,和最终的附加系统以辅助混合,例如上文已经描述的静态在线混合器或高剪切在线混合器。与在专用容器中混合相比,此类配置可特别减少设备投资和所需占地面积。调理混合器610的第二混合设备也可包括主动混合装置的此类专用容器和可能包括静态和/或高剪切在线混合器的管内注入系统的组合。或者,在步骤(a2)处,催化剂前体配制物104可以初始与20%的重质油原料101混合,所得混合重质油原料可与另外40%的重质油原料混合,以及所得60%的混合重质油原料可以根据逐步稀释的良好工程实践与剩余的40%的重质油混合,以便将催化剂前体配制物104彻底分散在重质油原料中。在本文中描述的适当混合装置或方法中的混合时间也应当用于逐步稀释方法。根据本发明的方法优选根据第二实施方案进行,其中步骤(a)包括(a1)将有机添加剂化合物102与催化剂前体组合物105预混合以产生催化剂前体配制物104,和(a2)将所述催化剂前体配制物104与所述重质油原料101混合。在步骤(a)处,重质油原料101与催化剂前体组合物104的混合可以对部分或全部的重质油原料101进行。根据一个或多个优选实施方案,混合步骤(a)在催化剂前体配制物104与送至加氢转化系统的重质油原料101的全部流之间进行。在一个或多个替代性实施方案中,混合步骤(a)在催化剂前体配制物104与送至加氢转化的重质油原料101的流的一部分之间进行。由此,制备经调理的重质油原料103可以通过将所述重质油原料101的至少一部分流,例如所述重质油原料101的流的至少50重量%与催化剂前体配制物104混合来进行。一旦已经加入催化剂前体配制物104,则可以再并入所述重质油原料101的流的补充部分,即在步骤(b)处将其预热之前与经调理的重质油原料103混合。步骤(b):加热经调理的重质油原料在步骤(a)中形成的经调理的油原料103随后在至少一个预热装置630中加热,随后引入混合床反应器进行加氢转化。将经调理的油原料103送至至少一个任选通过泵加压的预热装置630。预热装置包括本领域技术人员已知能够加热重质油原料的任何加热装置。该预热装置可包括至少包括预热室的炉、和/或油进料在其中流动的管、经调理的油原料与h2的混合器、任何类型的合适的热交换器,例如油进料在其中流动的管或螺旋热交换器等。这种经调理的重质油原料的预热允许在随后的步骤(d)中在混合加氢转化反应器中达到目标温度。经调理的油原料103更优选地在预热装置630中加热到280℃至450℃、甚至更优选300℃至400℃、且最优选320℃至365℃的温度,特别是为了随后在步骤(c)处在加氢转化反应器中达到目标温度。预热装置的表面温度,例如炉或(一个或多个)热交换器的腔室或管的钢壳的表面温度可以达到400℃至650℃。在步骤(a)处将包含催化剂前体组合物105和有机添加剂102的催化剂前体配制物104与重质油原料101混合避免或减少了在这些高温下在该预热装置中可能发生的结垢。根据一个或多个实施方案,将经调理的原料加热到比混合加氢转化反应器内的加氢转化温度低100℃、优选比加氢转化温度低50℃的温度。例如,对于410℃-440℃的加氢转化温度,可以在310℃-340℃的温度下在步骤(b)处加热经调理的油原料。绝对压力为大气压(例如0.101325mpa)至38mpa、优选5mpa至25mpa且优选6mpa至20mpa。在该步骤(b)中的加热有利地导致经调理的油原料释放硫,所述硫可以与催化剂前体组合物的金属结合。根据一个或多个实施方案,在预热装置630中加热的步骤(b)处在经调理的重质油原料中原位形成或至少开始形成胶体或分子催化剂。为了形成胶体或分子催化剂,硫必须可用(例如如h2s)以便与来自催化剂前体组合物的金属结合。在经调理的重质油原料中原位形成胶体或分子催化剂下面详细描述了在经调理的重质油原料中胶体或分子催化剂的原位一般形成,以及在步骤(b)和/或(c)处此类形成所需的条件。在其中重质油原料包含充足或过量的硫的情况下,可以通过将经调理的重质油原料103加热到足以从中释放硫的温度而原位形成最终的活化催化剂。硫源由此可以是溶解在重质油原料中的h2s,或包含在用于加氢转化的再循环到混合床加氢转化反应器中的氢气中的h2s,或来自存在于原料中或预先在重质油原料中最终引入(注入二甲基二硫化物、硫代乙酰胺、硫醇类型的任何含硫烃原料、硫化物、含硫石油、含硫瓦斯油、含硫减压馏分油、含硫渣油)的有机硫分子的h2s,此类注入是稀少的且针对高度非典型的重质油原料。硫源由此可以是原料中的硫化合物或添加到原料中的硫化合物。根据一个或多个实施方案,分散的胶体或分子催化剂的形成在大气压至38mpa、优选5至25mpa、且优选6至20mpa的绝对压力下进行。由于在步骤(a)处的彻底混合,在与硫反应以形成金属硫化物化合物时,可以形成分子分散的催化剂。在一些情况下,可能发生少量的附聚,产生胶体尺寸的催化剂颗粒。但是,据信在步骤(a)处小心地使前体组合物遍布重质油原料彻底混合将产生单独的催化剂分子而不是胶体颗粒。简单共混(虽然不能充分混合)通常导致形成微米尺寸或更大的大附聚金属硫化物化合物。为了形成金属硫化物催化剂,经调理的原料103优选加热到室温(例如15℃)至500℃、更优选200℃至500℃、甚至更优选250℃至450℃、且甚至更优选300℃至435℃的温度。在步骤(b)和/或(c)处使用的温度允许形成金属硫化物催化剂。由此,在步骤(c)处将加热的经调理的原料106引入混合床加氢转化反应器中之前,在该加热步骤(b)过程中,可由此(至少部分)形成胶体或分子催化剂。胶体或分子催化剂也可以在步骤(c)处在混合床加氢转化反应器自身内原位形成,尤其是在其已经在步骤(b)中开始形成的情况下完全或部分形成。经调理的油原料中钼的浓度优选为重质油原料101的按重量计的5ppm至500ppm、更优选按重量计的10ppm至300ppm、更优选按重量计的10ppm至175ppm、甚至更优选按重量计的10ppm至75ppm、且最优选按重量计的10ppm至50ppm。当挥发性馏分从非挥发性渣油馏分中除去时,mo可以变得更浓。由于胶体或分子催化剂往往是非常亲水的,单个颗粒或分子将倾向于向重质油原料中更亲水的部分或分子、尤其是沥青质迁移。尽管催化剂化合物的高极性性质导致或允许胶体或分子催化剂与沥青质分子缔合,但高极性催化剂化合物与疏水性重质油原料之间的一般不相容性使得必须在形成胶体或分子催化剂之前使油溶性催化剂前体配制物在重质油原料中精细或彻底混合。优选地,该胶体或分子催化剂包含二硫化钼。理论上,二硫化钼的纳米尺寸晶体具有夹在14个硫原子之间的7个钼原子,并且由此可用于催化活性的在边缘处暴露的钼原子总数大于二硫化钼的微米尺寸晶体中的钼原子总数。实际上,形成具有增强的分散的本发明中的小催化剂颗粒(即胶体或分子催化剂)导致更多催化剂颗粒和遍布该油原料更均匀分布的催化剂位点。此外,纳米尺寸或更小的二硫化钼颗粒被认为与沥青质分子紧密缔合。步骤(c):加热的经调理的原料的加氢转化随后将加热的经调理的原料106(任选通过泵加压,尤其是如果在步骤(b)之前尚未加压)与氢气601一起引入至少一个混合沸腾-夹带床反应器640中,并且在加氢转化条件下操作以产生提质的材料107。如前所述,如果在步骤(b)中未完全形成或根本没形成,则胶体或分子催化剂可以在步骤(c)处在混合床加氢转化反应器自身中原位形成。当胶体或分子催化剂在步骤(b)处在经调理的重质油原料中原位形成时,当进入至少一个混合沸腾-夹带床反应器640时,加热的经调理的原料106已经含有部分或全部的胶体或分子催化剂。该混合沸腾-夹带床反应器640包含固相、液态烃相和气相,所述固相包含膨胀床形式的多孔负载催化剂,所述液态烃相包含含有分散在其中的胶体或分子催化剂的所述加热的经调理的重质油原料106,且所述气相包含氢气。该混合沸腾-夹带床反应器640是沸腾床加氢转化反应器,除了保持在该沸腾床反应器中的膨胀床形式的多孔负载型催化剂之外,其包括与流出物(提质原料)一起夹带出反应器的分子或胶体催化剂。根据一个或多个实施方案,混合床加氢转化反应器的操作基于用于h-oiltm方法的沸腾床反应器的操作,例如描述在专利us4521295或us4495060或us4457831或us4354852中,或描述在论文aiche,1995年3月19-23日,houston,texas,论文编号46d,“secondgeneration ebullated bed technology”中。在该实施方式中,沸腾床反应器可包括再循环泵,其使得可以通过在反应器顶部抽出并在反应器底部再注入的至少一部分液体级分的连续再循环而将多孔负载固体催化剂保持为沸腾床(bubbling bed)。混合床反应器优选包括位于混合床反应器底部或其附近的输入口,通过该输入口将加热的经调理的原料106与氢气601一起引入,以及位于反应器顶部或其附近的输出口,通过该输出口排出提质的材料107。混合床反应器进一步包括包含多孔负载催化剂的膨胀催化剂区。混合床反应器还包括位于膨胀催化剂区下方的下部无负载催化剂区和位于膨胀催化剂区上方的上部无负载催化剂区。胶体或分子催化剂在混合床反应器(包括膨胀催化剂区和无负载催化剂区)中遍布该原料分散,由此可用于促进在常规沸腾床反应器中构成无催化剂区的区域内的提质反应。混合床反应器中的原料通过与沸腾泵连通的再循环通道从上部无负载催化剂区连续循环到下部无负载催化剂区。在再循环通道的顶部是漏斗形的再循环杯,通过该再循环杯从上部无负载催化剂区抽取原料。内部再循环原料与新鲜加热的经调理的原料106和补充的氢气601共混。如已知且例如专利fr3033797中所述,当多孔负载加氢转化催化剂用废时,可以通过优选在反应器底部取出废催化剂,并通过在反应器顶部或底部引入新鲜催化剂来用新鲜催化剂部分替换该多孔负载加氢转化催化剂。废催化剂的这种替换优选以规则的时间间隔进行,并且优选分批或基本上连续地进行。这些取出/替换使用有利地能使这种加氢转化步骤连续运行的装置进行。例如,通向膨胀催化剂区的输入和输出管可以分别用于引入/取出新鲜负载催化剂和废负载催化剂。在混合床反应器中存在胶体或分子催化剂在膨胀催化剂区、再循环通道以及下部和上部无负载催化剂区中提供了附加的催化加氢活性。自由基在多孔负载催化剂外部的封端使沉积物和焦炭前体的形成最小化,沉积物和焦炭前体通常是使负载催化剂失活的原因。这可以允许减少进行期望的加氢处理反应将另外所需的多孔负载催化剂的量。也可以降低必须取出和补充多孔负载催化剂的速率。在加氢转化步骤(c)中使用的加氢转化多孔负载催化剂可以含有沉积在载体上的一种或多种元素周期表第4至12族的元素。多孔负载催化剂的载体可以有利地为无定形载体,如二氧化硅、氧化铝、二氧化硅/氧化铝、二氧化钛或这些结构的组合,且非常优选为氧化铝。该催化剂可以含有至少一种选自镍和钴的第viii族金属,优选镍,所述第viii族元素优选与至少一种选自钼和钨的第vib族金属组合使用;优选地,第vib族金属是钼。在本说明书中,化学元素的族可以根据cas分类给出(crc handbook ofchemistry and physics,由crc press出版,editor in chief d.r.lide,第81版,2000-2001)。例如,根据cas分类的第viii族对应于根据新iupac分类的第8、9和10列的金属。有利地,在加氢转化步骤(d)中使用的加氢转化多孔负载催化剂包含氧化铝载体和至少一种选自镍和钴的第viii族金属、优选镍,和至少一种选自钼和钨的第vib族金属、优选钼。优选地,该加氢转化多孔负载催化剂包含镍作为第viii族元素,和钼作为第vib族元素。来自第viii族非贵金属的金属、特别是镍的含量以金属氧化物(特别是nio)重量表示有利地为0.5重量%至10重量%、和优选1重量%至6重量%,并且来自第vib族的金属、特别是钼的含量以金属氧化物(特别是三氧化钼moo3)重量表示有利地为1重量%至30重量%、和优选4重量%至20重量%。金属的含量表示为金属氧化物相对于多孔负载催化剂重量的重量百分比。这种多孔负载催化剂有利地以挤出物或珠粒的形式使用。该珠粒可具有例如0.4mm至4.0mm的直径。该挤出物具有例如直径为0.5mm至4.0mm和长度为1mm至5mm的圆柱体形式。挤出物也可以是不同形状的物体,如三叶形、规则或不规则的四叶形或其它多叶形。也可使用其它形式的多孔负载催化剂。这些各种形式的多孔负载催化剂的尺寸可以用当量直径来表征。当量直径定义为颗粒体积与颗粒外表面积之间的比率乘以六。以挤出物、珠粒形式或其它形式使用的多孔负载催化剂由此具有0.4mm至4.4mm的当量直径。这些多孔负载催化剂是本领域技术人员公知的。在加氢转化步骤(c)中,所述加热的经调理的原料106通常在用于重质油原料的加氢转化的常规条件下转化。根据一个或多个实施方案,加氢转化步骤(c)在2至38mpa、优选5至25mpa和优选6至20mpa的绝对压力下和在300℃至550℃、优选350℃至500℃、优选370℃至450℃、更优选400℃至440℃、且甚至更优选410℃至435℃的温度下进行。根据一个或多个实施方案,原料相对于每个混合反应器的体积的液体时空速(lhsv)为0.05h-1至10h-1、优选0.10h-1至2h-1且优选0.10h-1至1h-1。根据一个或多个实施方案,lhsv为0.05h-1至0.09h-1。lhsv定义为室温和大气压(通常为15℃和0.101325mpa)下每反应器体积的液体进料体积流速。根据一个或多个实施方案,与重质油原料106混合的氢气的量优选为每立方米(m3)液体重质油原料50至5000标准立方米(nm3),如100至3000nm3/m3和优选200至2000nm3/m3。根据一个或多个实施方案,加氢转化步骤(c)在一个或多个混合床加氢转化反应器中进行,所述混合床加氢转化反应器可以是串联和/或并联的。步骤(d):来自加氢转化步骤(c)的提质的材料的进一步处理可以进一步处理提质的材料107。此类进一步处理的实例包括但不限于以下中的至少一个:提质的材料的烃馏分的分离,在一个或多个补充的混合沸腾-夹带床反应器或沸腾床反应器中的进一步加氢转化以产生进一步提质的材料,进一步提质的材料的烃馏分的分馏,提质的材料107的至少一部分或来自提质的材料或进一步提质的材料的分馏的重质液体馏分的脱沥青,提质的或进一步提质的材料在保护床中纯化以除去至少一部分胶体或分子催化剂和金属杂质。可以将可由提质的材料107产生的各种烃馏分送至精炼厂中的不同工艺,并且本文不描述关于这些后处理操作的细节,因为它们通常是技术人员已知的,并且将会毫无意义地使描述复杂化。例如,可以将气体馏分、石脑油、中间馏分油、vgo、dao送至加氢处理、蒸汽裂化、流化催化裂化(fcc)、加氢裂化、润滑油提取等等的过程,渣油(常压或减压渣油)也可以被后处理,或用于其它应用,如气化、沥青生产等。重质馏分(包括渣油)也可以在加氢转化过程中、例如在混合床反应器中再循环。根据一个或多个实施方案,如图6中所示,该方法进一步包括:-由加氢转化步骤(c)产生的提质材料的至少一部分或全部或任选的由分离加氢转化步骤(c)产生的提质材料的一部分或全部的任选分离步骤产生的主要在大于或等于350℃的温度下沸腾的液体重质馏分603在氢气604的存在下在第二混合沸腾-夹带床反应器660中的第二加氢转化步骤,所述第二混合沸腾-夹带床反应器660包含第二多孔负载催化剂并且在加氢转化条件下操作以产生具有减少的重质渣油馏分、减少的康拉逊残炭和可能的减少量的硫和/或氮和/或金属的加氢转化液体流出物605;-在分馏段670中分馏所述加氢转化液体流出物605的一部分或全部以产生主要在大于或等于350℃的温度下沸腾的至少一种重质馏分607的步骤,所述重质馏分含有在大于或等于540℃的温度下沸腾的残余馏分;-在脱沥青器680中用至少一种烃溶剂将所述重质馏分607的一部分或全部脱沥青以产生脱沥青油dao 608和残余沥青609的任选步骤。所述第二加氢转化步骤以类似于对加氢转化步骤(c)所述的方式进行,且因此在此不再重复其描述。除下文给出的说明外,这尤其适用于操作条件、所用设备、所用加氢转化多孔负载催化剂。至于加氢转化步骤(c),第二加氢转化步骤在类似于混合床反应器640的第二混合沸腾-夹带床反应器660中进行。在该附加的加氢转化步骤中,操作条件可以与加氢转化步骤(c)中的那些相似或不同,温度保持为300℃至550℃、优选350℃至500℃、更优选370℃至450℃、更优选400℃至440℃、且甚至更优选410℃至435℃,并且引入反应器的氢气量保持为50至5000nm3/m3液体原料、优选100至3000nm3/m3、且甚至更优选200至2000nm3/m3。其它压力和lhsv参数在与对加氢转化步骤(c)所述那些相同的范围内。第二混合床反应器660中所用的加氢转化多孔负载催化剂可以与混合床反应器640中所用的相同,或者还可以是也适于重质油原料的加氢转化的另一多孔负载型催化剂,如对加氢转化步骤(c)中所用的负载催化剂所限定的。在分离段650中进行任选的分离步骤,分离提质的原料107的一部分或全部以产生至少两种馏分,包括主要在大于或等于350℃的温度下沸腾的重质液体馏分603。其它馏分602是(一种或多种)轻质馏分和中间馏分。由此分离的轻质馏分主要含有气体(h2、h2s、nh3和c1-c4)、石脑油(在低于150℃的温度下沸腾的馏分)、煤油(在150℃至250℃下沸腾的馏分)和至少一部分柴油(在250℃至375℃下沸腾的馏分)。随后可以将该轻质馏分至少部分送至分馏单元(图6中未图示),在那里例如通过穿过闪蒸罐从所述轻质馏分中提取轻质气体。由此回收的气态氢(其可以送至纯化和压缩设备)可以有利地再循环到加氢转化步骤(c)中。回收的气态氢也可以用于精炼厂的其它设备。该分离段650包括本领域技术人员已知的任何分离装置。其可以包括一个或多个串联布置的闪蒸罐,和/或一个或多个蒸汽和/或氢气-汽提塔,和/或常压蒸馏塔,和/或减压蒸馏塔,并优选由单个闪蒸罐构成,通常称为“热分离器”。分馏、分离来自第二加氢转化步骤的加氢转化液体流出物的一部分或全部以产生至少两种馏分的步骤在包括本领域技术人员已知的任何分离装置的分馏段670中进行,所述至少两种馏分包括主要在高于350℃、优选高于500℃和优选高于540℃的温度下沸腾的至少一种重质液体馏分607。(一种或多种)其它馏分606是(一种或多种)轻质馏分和中间馏分。重质液体馏分607含有在高于540℃的温度下沸腾的馏分,称为减压渣油(其是未转化的馏分)。其可以含有在250℃至375℃下沸腾的柴油馏分的一部分和称为减压馏分油的在375℃至540℃下沸腾的馏分。该分馏段670可包括一个或多个串联布置的闪蒸罐,和/或一个或多个蒸汽和/或氢气-汽提塔,和/或常压蒸馏塔,和/或减压蒸馏塔,并且优选由一组串联的多个闪蒸罐以及常压和减压蒸馏塔构成。当期望将一部分重质渣油馏分(例如一部分重质液体馏分607和/或一部分残余沥青609,或一部分dao 608)再循环回加氢转化系统(例如在混合床反应器640中或上游)时,可以有利地将胶体或分子催化剂留在渣油和/或残余沥青馏分中。可以对再循环料流进行清洗,通常用于防止一些化合物以过量水平积聚。本发明还涉及如上详述的被配置用于重质油原料101的加氢转化的沸腾-夹带床系统600。下面提及的附图标记涉及图6,其示意性图示了根据本发明的混合床加氢转化系统的实例。所述系统600包括:-调理混合器610,其被配置为通过将所述重质油原料101与催化剂前体配制物104混合来制备经调理的重质油原料103,所述催化剂前体配制物104包含含有钼的催化剂前体组合物105和有机添加剂,所述有机化合物102与钼的摩尔比为0.1:1至20:1;-至少一个预热装置630,其被配置为加热经调理的原料103;-至少一个混合沸腾-夹带床反应器640,其被配置为包括:--包含固相的膨胀催化剂床,所述固相包含多孔负载型催化剂作为固相,--包含加热的经调理的重质油原料106的液体烃相,所述重质油原料106含有分散在其中的胶体或分子催化剂;--和包含氢气的气相。所述至少一个混合沸腾-夹带床反应器640还被配置为在氢气的存在下并在加氢转化条件下操作,以便引起所述加热的经调理的原料中的烃的热裂解以提供提质的材料107。所述至少一个预热装置630和/或所述至少一个混合沸腾-夹带床反应器640还被配置为在所述经调理的重质油原料中形成胶体或分子催化剂。关于在沸腾-夹带床系统中使用的每个设备/装置/段的细节已经在上文中就方法给出并不再重复。实施例以下实施例图示了与根据现有技术的方法和系统相比根据本发明的方法和系统的一些性能品质,特别是减少的设备结垢,而不限制本发明的范围。该实施例基于使用称为alcor热液工艺模拟器或hlps的分析装置的测试,该分析装置模拟热交换器中常压渣油(ar)的结垢效应。在受控条件下将ar泵送穿过加热器管(层流管壳式热交换器)并且在加热器管上形成结垢沉积物。离开热交换器的ar的温度与沉积物对热交换器效率的影响相关。ar液体出口温度从其初始最大值的降低被称为δt,并与沉积量相关。δt的降低越高,结垢和沉积量越高。hlps测试可以用于通过比较相同测试条件下获得的ar液体出口温度的下降斜率来评价不同ar的结垢倾向。有机添加剂的有效性也可以通过比较纯样品(不含有机添加剂)与混合有有机添加剂的样品的测试结果来确定。测试两个样品:样品1是根据现有技术的重质油原料和分子或胶体催化剂的共混物,并且样品2是根据本发明的共混物,除了有机添加剂之外,其包含相同的重质油原料与相同的分子或胶体催化剂。所用的重质油原料(“进料”)是常压渣油(ar),其主要组成和性质在下表1中给出。[表1] 标准化方法 单位 进料 vgo(cpc稀释剂) 密度 nf en iso 12185 0.959 0.8677 ibp-350℃ astm d1160 重量% 21 2.7 350-540℃ astm d1160 重量% 35 95.5 540℃+ astm d1160 重量% 44 1.8 c astm d5291 重量% 84.5 86.5 h astm d5291 重量% 11.4 13.71 n astm d5291 重量% 0.3 0.0037 s nf iso 8754 重量% 3.81 0.074 ni astm d7260 重量ppm 25 <2 v astm d7260 重量ppm 78 <2 k astm d7260 重量ppm 2 <1 na astm d7260 重量ppm 196 <1 ca astm d7260 重量ppm <1 <1 p astm d7260 重量ppm <5 <5 si astm d7260 重量ppm <1 <1 fe astm d7260 重量ppm 3 6 ti astm d7260 重量ppm 79 <1 <![cdata[沥青质c<sub>5</sub>]]> uop99–07 重量% 10.6 0.2 <![cdata[沥青质c<sub>7</sub>]]> nf t60-115 重量% 4.7 0.05 康拉逊残炭 nf en iso 10370 重量% 11.3 0.2 样品1:样品1是进料(ar)和催化剂前体组合物(cpc)的共混物,该催化剂前体组合物是在减压瓦斯油(vgo)中稀释的2-乙基己酸钼。vgo的组成在上表1中给出。通过在70℃的温度下将2-乙基己酸钼与vgo混合并持续30分钟的时间段来获得cpc溶液。含有vgo的cpc溶液中钼含量为3500重量ppm。随后在70℃的温度下将cpc溶液与进料(ar)混合并持续30分钟的时间段。样品1中mo的含量为283重量ppm(参见下表2)。样品2:样品2是进料(ar)与和样品1相同的cpc溶液(用vgo稀释的2-乙基己酸钼)的共混物,并且其中有机添加剂为2-乙基己酸(2eha)。2eha的cas编号为149-57-5。首先将如对样品1详细描述的那样获得的cpc溶液与2eha在70℃的温度下混合并持续30分钟。随后,将含有有机添加剂2eha的cpc溶液与进料(ar)在70℃的温度下混合并持续30分钟。样品2中mo的含量为283重量ppm(参见下表2)。有机添加剂2eha的浓度为5761重量ppm(参见下表2)。2eha/mo的摩尔比=13.6。[表2]根据astm d7260测定样品中的mo含量。通过称重测定酸和酯有机添加剂含量。hlps测试条件在下表3中给出。[表3]不同样品的测试结果(样品1为s1,样品2为s2)显示在图7的曲线图中。x轴表示以小时为单位的时间,并且y轴表示在时间t处离开管的油共混物(样品)的温度[toil out]t与离开管的油共混物(样品)的最高温度[toil out]max之间的温度差δt:δt=[toil out]t-[toil out]max。结果表明,样品1具有强的结垢倾向,因为其δt迅速下降。根据本发明的含有有机添加剂如2eha的样品2具有比样品1更低的δt,表明在所述有机添加剂的作用下结垢行为显著降低。
背景技术:
0、现有技术
1、将重质油原料转化为有用的最终产物需要大量处理,包括降低重质油的沸点、提高氢/碳比、和除去杂质如金属、硫、氮和高碳形成化合物。
2、催化加氢转化通常用于重质油原料,并通常使用三相反应器来进行,其中使原料与氢气和催化剂接触。在该反应器中,催化剂可以以固定床、移动床、沸腾床或夹带床的形式使用,例如如2011年由technip出版的书籍“heavy crude oils:from geologyto upgrading,an overview”的第18章“catalytic hydrotreatment andhydroconversion:fixed bed,moving bed,ebullated bed and entrained bed”中所述。在沸腾床或夹带床的情况下,该反应器包括液体和气体的上升流。技术的选择通常取决于待处理的原料的性质,且特别是其金属含量、其对杂质的耐受性和目标转化率。
3、一些重质原料加氢转化方法基于混合使用不同的催化剂床类型的混合技术,例如使用沸腾床和夹带床技术、或固定床和夹带床技术的混合方法,由此通常充分利用每种技术。
4、例如,本领域已知在同一加氢转化反应器中当前使用保持在反应器中的沸腾床中的负载催化剂和与流出物一起夹带出反应器的较小尺寸的夹带催化剂,通常也称为“浆料”催化剂。第二催化剂的这种夹带特别通过该浆料催化剂的合适密度和合适粒度来实现。因此,“混合沸腾-夹带床”方法,在本文中也称为“混合沸腾床”或简称为“混合床”方法,在本说明书中限定为是指除了保持在沸腾床中的负载催化剂之外还包括夹带催化剂的沸腾床的实施方式,其可以视为沸腾床和夹带床的混合操作。该混合床在某种程度上是具有必然不同的颗粒尺寸和/或密度的两种类型的催化剂的混合床,一种类型的催化剂保持在反应器中,而另一种类型的催化剂——浆料催化剂——与流出物一起夹带出反应器。
5、已知此类混合床加氢转化方法用于改进传统的沸腾床方法,特别是由于添加夹带催化剂减少了加氢转化反应器系统中沉积物和焦炭前体的形成。
6、实际上,已知的是,在用于提质重质油的沸腾床反应器的操作过程中,将重质油加热到通常具有高分子量和/或低氢/碳比的重质油原料的高沸点馏分(其一个实例是统称为“沥青质”的一类复杂化合物)倾向于经历热裂化以形成链长减小的自由基的温度。这些自由基具有与其它自由基、或与其它分子反应以产生焦炭前体和沉积物的潜力。在反应器已经包含保持在反应器中的负载催化剂的同时,从底部到顶部穿过反应器的浆料催化剂提供了附加的催化加氢活性,尤其在反应器的通常不含负载催化剂的区域中。浆料催化剂因此在这些区域中与自由基反应,形成稳定的分子,并由此有助于控制和减少沉积物和焦炭前体的形成。由于焦炭和沉积物的形成是常规催化剂失活和加氢转化装置结垢的主要原因,此类混合方法能够提高负载催化剂的寿命并防止下游设备如分离容器、蒸馏塔、热交换器等的结垢。
7、例如,pct申请wo2012/088025描述了使用沸腾床技术和包含负载催化剂与浆料催化剂的催化体系来提质重质原料的此类混合方法。沸腾床反应器包含具有不同特征的两种类型的催化剂,第一催化剂具有大于0.65mm的尺寸并占据膨胀区,而第二催化剂具有1至300μm的平均尺寸并悬浮使用。第二催化剂与进料一起引入沸腾床,并从底部至顶部穿过该反应器。其或者由非负载的本体催化剂制备,或者通过压碎负载催化剂(粒度为1至300μm)制备。
8、专利文献us2005/0241991还涉及用于重质油的此类混合床加氢转化方法,并公开了一个或多个沸腾床反应器,其可以在原料中添加分散的有机可溶性金属前体的情况下以混合模式操作。在将其引入第一沸腾床反应器或后续沸腾床反应器之前,在与原料的精细混合阶段中进行催化剂前体的添加以制备经调理的原料,所述催化剂前体可在减压瓦斯油(vgo)中预先稀释。具体而言,该催化剂前体(通常为2-乙基己酸钼)一旦加热就通过与来自原料的加氢脱硫的h2s反应形成胶体或分子催化剂(例如分散的硫化钼)。此类方法抑制焦炭前体和沉积物的形成,否则这些焦炭前体和沉积物可能使负载催化剂失活并在沸腾床反应器和下游设备中结垢。
9、申请人的欧洲专利申请ep3723903也公开了用于重质油的混合床加氢转化方法,其中分散的固体催化剂获自结合钼与至少一种选自钴和镍的金属的strandberg、keggin、缺位keggin或取代缺位keggin结构的杂多阳离子的至少一种盐,从而改善加氢脱沥青并致使减少沉积物的形成。
10、用于重质油加氢转化的浆料催化剂,且特别是通过使用可溶性催化前体形成的胶体或分子催化剂,在本领域中是公知的。特别已知的是,某些金属化合物,如有机可溶性化合物(例如在us4244839、us2005/0241991、us2014/0027344中提及的环烷酸钼或辛酸钼)或水溶性化合物(例如在专利us3231488、us4637870和us4637871中提及的磷钼酸;在专利us6043182中提及的七钼酸铵;在fr3074699中提及的杂多阴离子的盐)可以用作分散催化剂前体并形成催化剂。在水溶性化合物的情况下,通常将分散的催化剂前体与原料混合以形成乳液。任选由钴或镍促进的分散催化剂(通常为钼)前体在酸性介质(在h3po4的存在下)或碱性介质(在nh4oh的存在下)中的溶解已经是许多研究和专利的主题。
11、除了可能出现在混合床反应器和下游设备中的焦炭前体和沉积物引起的结垢外,本发明人还观察到,一旦在将含有催化剂前体的重质油原料引入加氢转化反应器之前将其加热,结垢也可能出现在上游设备中。
12、在加氢转化反应器上游的设备中,尤其是在与特定胶体或分子催化剂的催化剂前体混合的重质油原料的加热设备中的此类结垢似乎主要与壁上的金属和碳的积聚有关,并可以限制设备的可操作性。
13、由此,尽管在已知的混合方法中的浆料催化剂(如上述那些)已知减少加氢转化反应器和下游设备中由焦炭前体和沉积物引起的结垢,但在含有与催化剂前体混合的重质油原料的上游设备中,如在预热装置中观察到的结垢构成了迄今尚未解决的另一操作问题。此外,已经观察到,在一些情况下,在下游设备中仍可能发生由焦炭前体和沉积物引起的结垢,表明仍可改进添加浆料催化剂的性能。
技术实现思路
本文地址:https://www.jishuxx.com/zhuanli/20240726/129861.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。