一种中药汤剂中塑化剂的液相色谱检测方法
- 国知局
- 2024-08-05 12:16:03
本发明属于医药检测,具体涉及中药汤剂中塑化剂的检测方法。
背景技术:
1、中药汤剂是将中药饮片用煎煮或浸泡去渣浓缩取汁的方法得到的液体剂型,是中药制剂中历史最悠久、应用最广泛的一种剂型[1]。由于中药汤剂能适应中医辨证论治、随证灵活加减的需要,中药汤剂内服和/或外用成为中医治疗疾病的主要手段之一[2]。随着社会发展,中药代煎汤剂以其方便、快捷、专业、经济等方面优势,越来越受到患者青睐[3],同时,中药代煎汤剂的质量控制与管理也日益受到业内的重视[4]。
2、邻苯二甲酸酯类塑化剂(phthalate esters,paes)是邻苯二甲酸酐与醇形成的酯的统称,为无色油状粘稠液体,易溶于有机溶剂,难溶于水,与pvc/pp/pe等塑料分子的相溶性较好,通过氢键或范德华力作用,当塑料制品接触到食品/药品中所含的水、油脂等时,其中的塑化剂易发生迁移进入液体中[5-6]。由于paes的脂溶性特点,其进入人体后,易在血液和组织(如肝脏、肾脏和肺)中蓄积[7],其毒性反应具有剂量、时间、年龄相关性,对孕妇、儿童和老年人的潜在危险较高。中国、美国、欧盟、日本等国家和地区均对paes的使用作了法律法规的限制。由于我国paes的使用量占全球塑化剂总用量的80%以上[8-9],限量塑化剂的检测和控制显得尤为重要。目前,中药代煎质量标准中无paes相关要求,但在我国国家强制标准《食品接触材料及制品用添加剂使用标准》(gb9685-2016)中对几种paes作了最大使用量、最大残留量(qm)、特定迁移限量(sml)的规定;其中,邻苯二甲酸二(2-乙基)己酯(dehp)的sml≤1.5mg/kg,邻苯二甲酸二烯丙酯(dap)的sml≤0.01mg/kg,邻苯二甲酸二异壬酯(dinp)的sml≤9mg/kg,邻苯二甲酸二正丁酯(dbp)的sml≤0.3mg/kg;此外适用于可被放入口中的所有玩具和儿童护理品的《玩具用涂层中有害物质限量》(gb24613-2009)有限定:dehp、dbp、bbp(邻苯二甲酸丁基苄酯)三者最大使用量总和≤0.1%,dinp、didp(邻苯二甲酸二异癸酯)、dnop(邻苯二甲酸二正辛酯)三者最大使用量总和≤0.1%。
3、鉴于中药汤剂的成分复杂,涉及的药物种类及其杂质来源多样,本发明课题组围绕上述国家强制标准中7种限量塑化剂(dehp、dap、dbp、bbp、dinp、didp、dnop)展开实验研究,建立一种准确方便的paes含量测定方法。
4、文献调查发现,大量研究对paes的检测采用气质联用色谱法(gc-ms)[10-11]、高效液相色谱法(hplc)[12-13];gc-ms法中dinp干扰性高,需要单独检测;而hplc法有望对paes进行一次性检测。因此,本研究选用hplc法,并从萃取、检测时间、分离度进行优化,讨论不同前处理方法的提取效率,并与gc-ms检测结果进行复核,最终建立运用hplc检测中药汤剂中7种邻苯二甲酸酯类塑化剂(dehp、dap、dbp、bbp、dinp、didp、dnop)含量的技术方案。
技术实现思路
1、本发明的目的在于提供一种提取、分离、并一次性检测中药汤剂中塑化剂的方法,其分离度高、重复性好,满足gb9685-2016中对相关paes的检测限要求。
2、本发明提供的中药汤剂中塑化剂的检测方法,采用液相色谱法,主要针对中药代煎汤剂中塑化剂的检测,具体步骤如下:
3、(一)配制7种邻苯二甲酸酯类(paes)系列标准品溶液:
4、取7种paes标准品:
5、邻苯二甲酸二烯丙酯(dap,如c16169000,质量浓度97.0%),邻苯二甲酸二(2-乙基)己酯(dehp,如c13342300,质量浓度99.0%),邻苯二甲酸二异壬酯(dinp,如c16174000,97.0%),邻苯二甲酸二正丁酯(dbp,如c16171000,质量浓度99.0%),邻苯二甲酸丁基苄酯(bbp,如c16168000,质量浓度97.0%),邻苯二甲酸二正辛酯(dnop,如c16175000,质量浓度97.5%);邻苯二甲酸二异癸酯(didp,如c16173550,99.0%);甲醇作为溶剂;
6、配制7种paes标准品溶液:
7、称取上述7种paes标准品各10.0mg,并置于同一根具塞试管中,加入甲醇,定容至100.0ml,超声振荡,得100.0mg/l的混合标准品溶液,置于冰箱0~5℃避光保存;
8、取所述混合标准品溶液1.0ml,再加甲醇定容至10.0ml;依次逐级稀释,配制得浓度梯度分别为0.2mg/l、0.5mg/l、1.0mg/l、5.0mg/l、10.0mg/l、20.0mg/l、50.0mg/l、100.0mg/l,共计8份系列标准品溶液样品,供hplc检测分析用。
9、(二)建立paes系列标准品溶液的hplc检测条件:
10、采用高效液相色谱仪(thermo vanquish)、二极管阵列检测器(thermo vanquish)
11、设定色谱条件:选用色谱柱agilent zorbax sb-c18;进样量:10μl;流速:1.0ml/min;柱温:30℃;紫外检测波长:240nm;
12、设定梯度洗脱程序:流动相包括流动相a和流动相b;设定参数为:流动相a为甲醇和乙腈混合溶液,甲醇:乙腈=1:1;流动相b为超纯水;
13、a相梯度参数预设置为:0~10min,40~80%;11~20min,80%;21~30min,80~100%;31~45min,100%;45~50min,100~40%。
14、(三)建立色谱峰面积与浓度之间关系的回归方程:
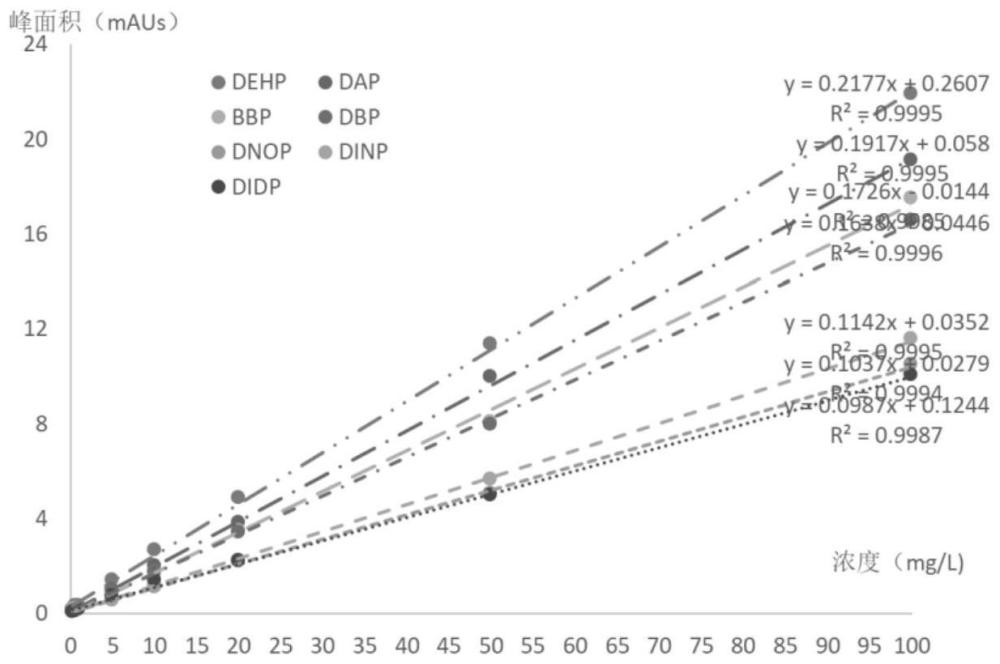
15、根据上述hplc检测条件,将8份系列标准品溶液样品依次进样检测,如图1所示,7种邻苯二甲酸酯类塑化剂dehp、dap、dbp、bbp、dinp、didp、dnop分离度良好,无杂志干扰;每个浓度进样3次,以3次峰面积的平均值为纵坐标(y),浓度为横坐标(x),如表1;绘制标准曲线,建立回归方程:
16、y = ax + b; (1)
17、a,b为线性方程系数;如表2所示;
18、表1,7种paes的平均峰面积(mau)与浓度(mg/l)表
19、
20、如表2所示,7种paes在0.2~100mg/l范围内具有较好的线性关系;另外,按信噪比(s/n)=3计算各paes的检出限,按s/n=10计算定量限;
21、表2,7种paes的回归方程、线性范围、相关系数、检出限、定量限
22、
23、精密度实验:取对照品溶液1ml,按前文色谱条件,重复进样6次,测得峰面积,计算rsd为1.37%~2.99%,表明本实验方案精密度较好。
24、(四)中药代煎汤剂的hplc定量检测
25、(1)中药代煎汤剂样品的前处理
26、任取中药代煎汤剂一份,剪开1袋,将中药煎液倒入烧杯内,盖上玻璃板。准确量取中药代煎汤剂2.0ml于具塞玻璃管中,边搅拌边加入甲醇8.0ml,超声振荡10min,2000r/min离心5min,过12.5cm定性滤纸滤去沉淀,再过0.45μm微孔滤膜。重复以上步骤,得50.0ml样品溶液,备用。此时稀释倍数为20%,利用氮吹仪挥发甲醇浓缩溶液至10.0ml,得到与原浓度一致的样品溶液1份,检测时视检测限必要时可加甲醇再度稀释。
27、(2)中药代煎汤剂的hplc定量分析
28、采用步骤(二)所述hplc检测条件,将中药代煎样品溶液依次进样检测,记录色谱峰保留时间、峰面积;按回归方程(1)中对应于dehp、dap、dbp、bbp、dinp、didp、dnop的7个回归方程,根据对应的峰面积(y),计算得到各paes浓度(x)。
29、本发明实施例中,给出了6例采用本发明检测的中药代煎汤剂样品溶液中paes的hplc图谱及对应的paes浓度值。
30、中药在代煎制备过程中,应尽量避免中药与塑料制品接触,如果无法避免与塑料制品接触,则应近量缩短接触时间、降低接触温度。例如煎煮前的中药浸泡过程,采用不锈钢桶;中药煎煮容器使用前后用温水清洗干净;煎煮用水直接取自自来水龙头;包装中药代煎液使用符合相关产品标准的中药代煎包装袋;机器煎煮完成后降温并灌装,放冷至常温后,置于冰箱5℃保存。
31、在实验过程中,本发明通过实验组和对照组进行对比分析。
32、抽取中药代煎处方50张,调配7帖,按上述要求代煎并包装成200ml/袋*14袋,放冷后放置于冰箱5℃冷藏。
33、实验组——煎液样品溶液的制备:剪开中药代煎汤包,将中药煎液倒入烧杯内,盖上玻璃板。准确量取中药代煎汤剂2.0ml于具塞玻璃管中,边搅拌边加入甲醇8.0ml,超声振荡10min,2000r/min离心5min,过12.5cm定性滤纸滤去沉淀,再过0.45μm微孔滤膜。重复以上步骤,得50.0ml样品溶液,备用。此时稀释倍数为20%,利用氮吹仪使甲醇挥发浓缩至10.0ml,得到与原浓度一致的样品溶液,检测时视检测限情况必要时可加甲醇再度稀释。
34、对照组——蒸馏水模拟液样品溶液的制备:将蒸馏水6l加至中药代煎机中煎煮2h,得约3l液体,放冷后包装成14袋,至常温后于冰箱5℃保存。取其中1袋,剪开,倒入烧杯内,量取2.0ml具塞玻璃管中,加甲醇定容至10.0ml,超声振荡10min,2000r/min离心5min,过0.45μm微孔滤膜。重复上述步骤,得50ml蒸馏水模拟液样品溶液,利用氮吹仪使甲醇挥发浓缩至10.0ml,得到与原浓度一致的样品溶液,备用。
35、对煎液样品组溶液进样检测,记录色谱峰保留时间、峰面积,根据紫外吸收光谱定性,根据回归方程计算浓度。
36、对于杂质干扰严重的样品,采用前期研究建立的气质联用色谱法[15]检测结果进行比对。
37、加样回收实验:取1.0ml浓度为1.0mg/l的混合paes标准品溶液,加入一份已测的中药代煎液样品,定容至10.0ml,振荡混匀,根据前文所述色谱条件,重复进样6次检测,计算回收率,如表3所示,回收率为93.33%~103.25%,rsd为0.37%~4.15%,表明本实验方案重复性和回收率较好。
38、综上数据,本检测方案满足中药代煎汤剂中7种paes的限量检测要求。
39、表3,某份中药代煎液样品的paes加标回收实验
40、
41、本发明在实验过程中所用材料、工艺条件的选取:
42、(1)提取溶剂的选择:首先根据7种paes的化学性质,确立采用有机溶剂超声提取的方法,实验比较了甲醇、乙腈、乙醚、正己烷对paes标准品混合溶液和中药代煎汤剂的提取效率,7种paes均易溶于这几种溶剂。表4显示甲醇、乙腈、乙醚、正己烷对paes标准品混合溶液提取后进样计算的回收率。加入正己烷振荡提取1次后,正己烷萃取层能有效溶解标准品中的paes,但在溶解提取中药汤剂后,检测时发现干扰偏大,然而提取效率好,可作为备选项;用无水乙醚提取时,乙醚与中药汤剂不能充分互溶,在充分震荡,分层明显,经萃取1~3次后检测,3次萃取的回收率与正己烷相当,提取效率不高;用甲醇提取时,甲醇加入中药汤剂后,随着甲醇加入量的提高,溶液从互溶向部分沉淀过渡,文献[17-18]研究表明,中药汤剂中的醇沉现象,在一定程度上去除了煎液中淀粉、树胶、果胶、粘液质、蛋白质、鞣质、色素、无机盐等水溶性杂质,有利于后期的检测;用乙腈提取时,乙腈加入中药汤剂后,部分中药汤剂发生絮凝现象,经振荡后絮凝消失。由于乙腈毒性较大,且絮凝现象不能有效排除杂质干扰,因此乙腈不能作为主要溶剂用于提取中药代煎液。由于流动相采用了甲醇/乙腈的溶剂体系,综合起来,本发明选择甲醇作为paes标准品溶液和中药代煎液的提取剂。
43、表4,各溶剂萃取1次和3次后对paes标准品混合溶液的提取回收率
44、
45、(2)中药汤剂种paes的分离提取时甲醇加入量的选取:实验中发现加入甲醇可以使中药煎液产生少量沉淀,经上述色谱条件的hplc检测结果与前期研究使用的gc-ms结果比较,paes含量无明显差异,说明,醇沉不影响paes的检测。因此,一定量的甲醇有利于去除杂质干扰,有利于实验分析。实验比较了20%~80%的甲醇含量对沉淀生成的关系,具有明显的线性关系。甲醇的比例越高,产生的可滤除的沉淀越多,溶液静置的时间越短,且不受超声振荡的影响。本发明在中药汤剂处理时甲醇加入后浓度达到75%~80%,能有效去除中药代煎汤剂中的蛋白质、淀粉、色素、无机盐类等物质,有助于后期紫外吸收峰的分离。
46、(3)检测波长、色谱柱、柱温的选择:
47、设置检测器参数在195nm~400nm范围内,对7种paes进行波长扫描,结果显示:7种paes在228nm~286nm处有较高的特征吸收峰;其中,228nm处为bbp的最强吸收峰,238nm处为dehp的最强吸收峰,241nm~245nm处为dap和dbp的最强吸收峰,272nm~286nm处dinp、didp、dnop的吸收峰较高,考虑各个paes的特征吸收波长以及干扰情况,本发明选择240nm为检测波长,可获得7种paes的较理想吸收峰,且dap、dehp特征吸收峰较高,如图1所示。
48、根据被测paes的理化性质,选用酸碱性范围较宽的c18柱,以满足中药煎液中7种paes的分离。实验测试了柱温20~50℃对7种paes分离度的影响:20℃、30℃时,出峰时间需要10~18min,分离度好,完成7种paes的分离需要45~65min;40℃时,出峰时间在7min左右,25min内完成色谱图,但dnop、dinp的色谱峰相互干扰,无法良好的分离;50℃,dnop、dinp、didp的色谱峰均无法分离。综合温度对色谱柱的影响以及分离效果,实验设定柱温为30℃。
49、(4)流动相和梯度洗脱程序的设定:
50、在分离paes的研究中,本发明参考文献[19-20]以甲醇和乙腈等比例混合溶剂为流动相a,超纯水为流动相b,通过实验尝试得到有效分离7种paes的梯度洗脱程序,同时相对降低了基线对paes的影响,获得良好的检测效果。
51、a相梯度变化为:0~10min,40~80%;11~20min,80%;21~30min,80%~100%;31~45min,100%;45~50min,100%~40%。实验表明,甲醇/乙腈溶剂在0~10min内由40%提高到80%,对应的dap出锋时间在11~15min内,变化不明显;11~20min甲醇/乙腈溶剂保持在80%,能实现bbp和dbp的有效分离;在21~30min调整甲醇/乙腈溶剂由80%上升到100%,可得到dehp的最大吸收峰;31~45min甲醇/乙腈溶剂保持100%,能实现保留时间非常接近的dnop、和dinp有效分离,如图1所示。
本文地址:https://www.jishuxx.com/zhuanli/20240802/261875.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表