一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法与流程
- 国知局
- 2024-07-12 10:17:45
本发明属于微生物和计算机科学交叉的,具体涉及一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法。
背景技术:
1、随着基因测序技术的进步和测序成本不断下降,高通量、多维度的微生物组学研究激增,传统的统计方法已经不再适用于极度高维、稀疏的微生物组数据分析,而适用于复杂数据分析的机器学习逐渐成为微生物组学数据分析的首选方法。机器学习已被证明是分析微生物群落数据并对特定结果进行预测(包括人类和环境健康)的有效方法,基于微生物群落丰度数据的机器学习已被成功用于预测人类年龄、健康或疾病状态、环境质量和环境中污染的存在,并可以作为法医学中的微量证据。对于人类健康,能够根据机器学习的肠道微生物结果提供精准干预和个体化营养指导。基于微生物丰度数据的机器学习算法已经在肠道微生物、皮肤微生物、土壤微生物、植物微生物、环境微生物等领域应用广泛。
2、微生物数据输入是机器学习数据输入的第一步,现有输入形态为样本对应不同菌相对丰度表,一般经过相对丰度筛选和标准化等简单数据预处理,导入机器学习模型中进行训练,存在无法建模和预测结果不良等局限。比如,中国专利一种基于肠道菌群预测模型集成学习的年龄预测方法(cn 114093515 a)中采用对肠道微生物丰度数据先采用归一化处理,选择特征后利用一维向量进行计算或者最佳算法和年龄预测模型;文章(performance of gut microbiome as an independent diagnostic tool for20diseases:cross-cohort validation of machine-learning classifiers)仅过滤了低丰度(相对丰度<0.001)的分类群和z score标准化的预处理,就进行机器学习建模分类20种疾病。以上数据的形态都为一维向量,形式为[a1,a2,a3,…,an],当前主流的预处理(中心化、归一化、log10、z score标准化)并不改变数据形态;而且ai、aj(i、j∈n)间可能有一定关联性,不利于数据的特征提取。在此形态下一般采用多层感知机mlp进行建模,模型本身表达力有限,对复杂问题无法建模。
技术实现思路
1、为了弥补现有技术的不足,本发明旨在提供一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,将数据进行升维和分解为一组正交基的线性组合,这样既可以引入复杂模型建模,也有利于数据特征的分解和提取。解决了现有的微生物数据导入机器学习模型进行训练时存在无法建模和预测结果不良等局限性的问题进行优化。具体技术方案如下:
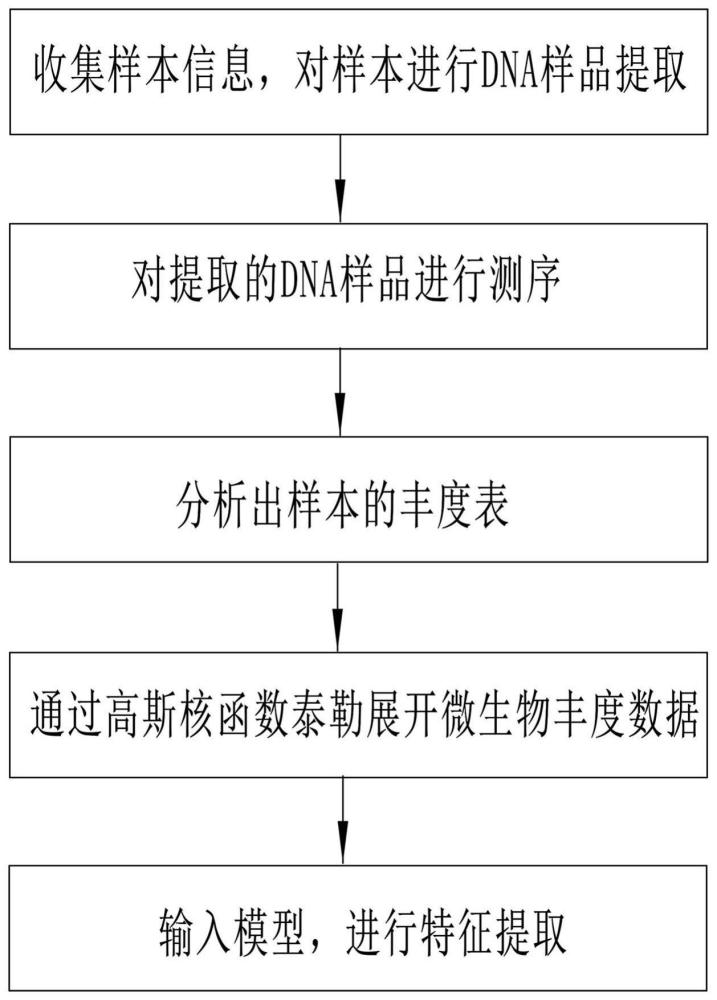
2、一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,所述方法具体步骤包括:
3、s1.收集样本信息,对样本进行dna样品提取;
4、s2.对提取的dna样品进行测序;
5、s3.分析出样本的丰度表;
6、s4.通过高斯核函数泰勒展开微生物丰度数据,得到一组正交基的线性组合;
7、s5.将s4中得到的一组正交基的线性组合输入模型,进行特征提取。
8、进一步地,所述步骤s2中将dna样品进行16s rrna基因测序分析,对rrna基因可变区v3+v4进行pcr扩增,细菌16s rrna的v3+v4区域通用引物:
9、正向引物338f(5’-actcctacgggaggcagcag-3’),
10、反向引物806r(5’-ggactachvgggtwtctaat-3’)。
11、进一步地,所述步骤s3中通过qiime2分析出丰度表,具体步骤包括:
12、s31.使用q2-tools插件中的import方法将下机数据集导入至qiime2平台中;
13、s32.用q2-demux的summarize方法将已经拆分好的样本进行信息统计和可视化处理,以获得序列数据中每个位置处序列质量分布的摘要;
14、s33.使用q2-dada2插件中集成的denoise-paired方法对双端序列进行降噪、合并、去嵌合体和去重复等处理,从而获得样本的代表性序列;
15、s34.探索样本的物种组成,并将其与样元数据再次组合,使用经过naive bayes分类器预训练的feature-classifier插件和在greengenes 13_8 99%otu上训练的分类器进行这项工作,使用qiime1将频率分布表转化为百分比数据,summarize_taxa.py命令按门、纲、目、科、属五个级别进行分类汇总,得到最后分类注释的菌属丰度表。
16、具体的,所述步骤s4中具体步骤包括:
17、s41.基于python的pytorch框架,将获得的肠道菌群物种的一维向量丰度表形式为[a1,a2,a3,…,an],取n=n,n根据实际样本个数而定;对其一条采样数据,把它形式化为一个向量:
18、v=[a0,a1,a2,...,ak]k∈n;
19、s42.对v进行第一次预处理,选用log10或crf标准化处理,使各指标值处于同一数量级别以进行综合测评分析:
20、x*=log10(x)/log10(max),
21、式中,x*为标准化后的数值,x为未标准化的原始丰度值,max为未标准化的最大原始丰度值。
22、s43.对进行了第一次预处理的数据通过高斯核函数泰勒展开方法,进行第二次预处理,得到升维数据。
23、具体的,所述步骤s43中利用高斯核函数的泰勒展开方法对一维向量进行嵌入处理,将向量变为[a1,a2,…,an]的二维矩阵形式:
24、
25、基于该高斯核函数公式对已经经过第一次预处理的数据ak进行升维,得到向量ak,a的维度为n;故得到
26、
27、或
28、即将数据分解为一组正交基的线性组合。
29、进一步地,所述步骤s5是基于python的pytorch框架,将步骤s4中得到的一组正交基的线性组合,引入到基于self-attention的encoder模型,进行特征提取。
30、进一步地,所述步骤s5具体包括:选取n层encoder,n∈[1,96],最后通过一层全连接层映射到预测空间;采取crossentropy loss计算距离,优化器选择随机梯度下降或自适应矩估计优化器进行参数更新;采用十倍交叉验证法,得到训练结果。
31、与现有技术相比,本发明有以下优点:本发明方法采用基于高斯核函数的泰勒展开方法,对一维向量进行嵌入处理,将数据分解为一组正交基的线性组合,不仅能够引入复杂模型建模,还有利于更加精准且全面的数据特征的分解和提取。
技术特征:1.一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,其特征在于,所述方法具体步骤包括:
2.根据权利要求1所述的一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,其特征在于,所述步骤s2中将dna样品进行16s rrna基因测序分析,对rrna基因可变区v3+v4进行pcr扩增,细菌16s rrna的v3+v4区域通用引物:
3.根据权利要求1所述的一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,其特征在于,所述步骤s3中通过qiime2分析出丰度表,具体步骤包括:
4.根据权利要求1所述的一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,其特征在于,所述步骤s4中具体步骤包括:
5.根据权利要求4所述的一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,其特征在于,所述步骤s43中利用高斯核函数的泰勒展开方法对一维向量进行嵌入处理,将向量变为[a1,a2,…,an]的二维矩阵形式:
6.根据权利要求1所述的一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,其特征在于,所述步骤s5是基于python的pytorch框架,将步骤s4中得到的一组正交基的线性组合,引入到基于self-attention的encoder模型,进行特征提取。
7.根据权利要求6所述的一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,其特征在于,所述步骤s5具体包括:选取n层encoder,n∈[1,96],最后通过一层全连接层映射到预测空间;采取crossentropy loss计算距离,优化器选择随机梯度下降或自适应矩估计优化器进行参数更新;采用十倍交叉验证法,得到训练结果。
技术总结本发明公开一种基于高斯核函数泰勒展开的微生物丰度数据嵌入方法,所述方法具体步骤包括:S1.收集样本信息,对样本进行DNA提取;S2.对提取的DNA样品进行测序;S3.分析出样本的丰度表;S4.通过高斯核函数泰勒展开微生物丰度数据,得到一组正交基的线性组合;S5.将S4中得到的一组正交基的线性组合输入模型,进行特征提取。本发明方法采用基于高斯核函数的泰勒展开方法,对一维向量进行嵌入处理,将数据分解为一组正交基的线性组合,不仅能够引入复杂模型建模,还有利于更加精准且全面的数据特征的分解和提取。技术研发人员:向沙沙,方智毅,苏晓明,王鑫洋,沈宇标,朱炫,陈杰受保护的技术使用者:杭州负熵健康科技有限公司技术研发日:技术公布日:2024/6/11本文地址:https://www.jishuxx.com/zhuanli/20240615/86025.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
上一篇
一种医用手套自动穿戴装置
下一篇
返回列表