拓扑蛋白质的程序化设计方法
- 国知局
- 2024-07-12 10:19:34
本发明涉及拓扑蛋白质合成领域,更具体地涉及拓扑蛋白质的程序化设计方法。
背景技术:
1、拓扑蛋白质是一类具有复杂化学拓扑结构的非线型蛋白质,为抗体工程、工业酶、生物材料等领域提供了崭新的研究对象,具有重要的基础科学意义和应用价值。受限于生命中心法则,细胞内的蛋白质合成遵循严格的模板合成机制,难以突破线型主链结构。天然拓扑蛋白质的生物合成往往涉及复杂的翻译后修饰过程,很多机理尚不明确,难以应用到人工拓扑蛋白质的设计与合成中。但借鉴大自然的理念,通过控制蛋白质链的空间关系,结合高效、特异的蛋白质化学反应,有望从线型蛋白质前体衍生出各种各样复杂的化学拓扑结构。
2、目前,人们已经开发出一些基于“组装-反应”协同的拓扑蛋白质合成策略,通过使用已知的蛋白质缠结基元和蛋白质反应基元来实现特定拓扑结构蛋白质(如纽结、链环等)的生物合成。但整体来看,蛋白质拓扑工程仍然处于初步阶段,这一方面是由于作为模板的蛋白质缠结基元非常有限,且难以对其进行改造和拓展;另一方面是由于蛋白质缠结基元和蛋白质反应基元的体积较大,导致在目标蛋白质中留下冗余基元,所述冗余基元既影响拓扑蛋白质构效关系的判断,又无法充分发挥化学拓扑结构的稳定化效果,使得问题变得更加复杂。人们亟待摆脱这些模板的限制,将已知的线型蛋白质结构重新设计,“无痕”改造为具有特殊拓扑结构的蛋白质,以求更大程度地发挥化学拓扑重塑构象空间的优势。
3、基于此,本发明致力于通过编辑目标蛋白质本身来引入分子链的缠结,实现“无痕”或“微痕”的拓扑改造策略,希望能够发展一个系统且普适的、程序化的拓扑蛋白质设计方法。
技术实现思路
1、发明要解决的问题
2、针对现有技术中存在的缠结基元种类少、体积大、易在实现目标拓扑结构的同时引入冗余基元等问题,本发明提供了一种拓扑蛋白质的程序化设计方法,通过编辑蛋白质自身二级结构之间的连接关系和空间关系,实现拓扑蛋白质的程序化设计。
3、用于解决问题的方案
4、本发明鉴于上述现有技术中存在的问题,进行了深入研究、反复试验,在保留目标蛋白质三级结构的基础上,通过改变二级结构基元间的连接关系引入分子链的缠结,设计出与目标蛋白质的三级结构和序列组成相似但是化学拓扑结构迥异的拓扑蛋白质变体,从而完成了本发明。即本发明如下所述:
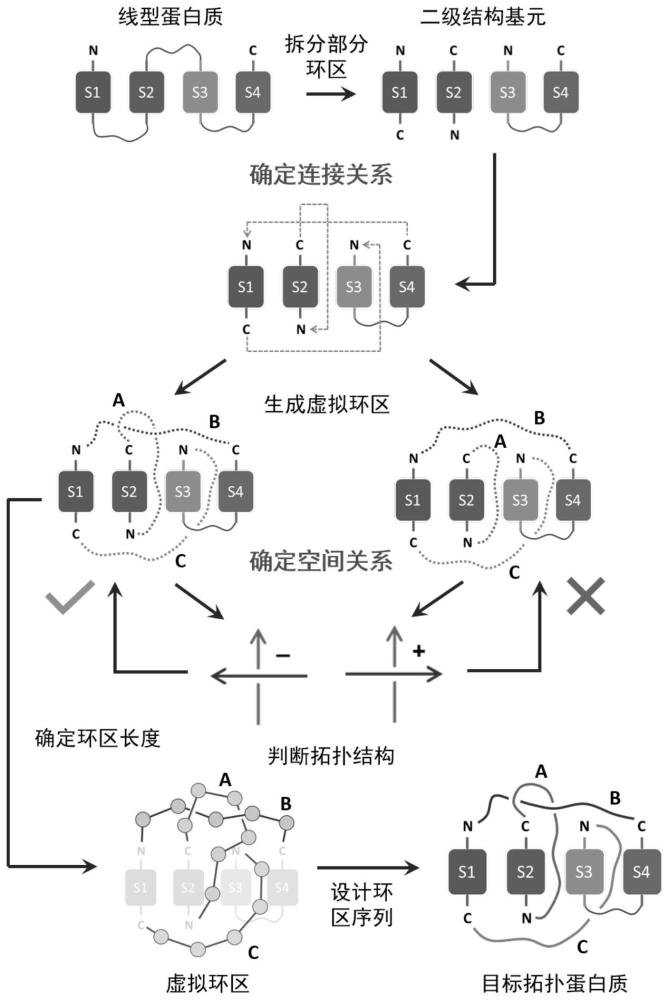
5、本发明在第一方面提供了一种拓扑蛋白质的程序化设计方法,所述方法包括以下步骤:
6、i)对目标蛋白质原有结构进行拆分,根据目标拓扑结构设计可能的重新连接方式;
7、ii)对结构基元间的连接方式进行评价,确定其在后续设计中的优先级;
8、iii)针对每种连接方式,依次生成新的虚拟环区,遍历所有可能的环区生成顺序组合,产生相应的空间关系,判断所形成的化学拓扑结构,计算目标拓扑结构的形成概率,并确定新生成的环区长度范围;
9、iv)设计拓扑蛋白质新生成的环区的长度和序列。
10、在一些具体的实施方式中,上述拓扑蛋白质的程序化设计方法中的步骤i)~iv)的具体操作如下:
11、i)以二级结构为基元拆分目标蛋白质原有的三级结构,被拆分环区的数目从两个开始依次增加,优先关注环区拆分数目少的方式,确定拆分方式后,根据目标蛋白质的化学拓扑结构,设计二级结构基元间的所有可能的新的连接方式;
12、ii)对步骤i)中设计的新的连接方式进行打分评价,并根据打分评价的结果确定不同的连接方式在后续拓扑结构设计中的优先级;
13、iii)根据步骤ii)中的打分评价的结果,按优先级顺序对每种连接方式进一步设计其空间关系,通过依次在每个待连接的位置生成新的虚拟环区,遍历所有可能的环区生成顺序的组合,产生相应的空间关系,判断生成新的虚拟环区后所得蛋白质的化学拓扑结构,并计算该连接方式下目标拓扑结构的形成概率,从而确定该连接方式下形成目标拓扑结构所对应的虚拟环区的相对空间关系以及虚拟环区的长度范围;
14、iv)根据步骤iii)确定的虚拟环区的相对空间关系和长度范围,从中优选实际环区的具体长度,并进一步设计其氨基酸序列,作为拓扑蛋白质新生成的环区序列,得到最终的拓扑蛋白质。
15、在具体实施方式中,步骤i)中所述的拆分为在环区处进行拆分;步骤i)中所述连接指的是在拆分之后的二级结构基元的n端和c端之间生成新的环区。
16、根据本发明在第一方面提供的拓扑蛋白质的程序化设计方法,步骤i)中所述目标蛋白质原有的三级结构中含有n个二级结构基元和n个环区,其中所述环区包括n端和c端之间的虚拟环区,当所设计的目标蛋白质拓扑结构为[2]索烃时,具体按照以下方法对所述目标蛋白质原有的三级结构进行拆分、确定新的连接方式:
17、①对目标蛋白质原有的三级结构中的2个环区进行拆分,共有n(n-1)/2种拆分方式和n(n-1)/2种新的连接方式;
18、②对目标蛋白质原有的三级结构中的3个环区进行拆分,共有n(n-1)(n-2)/6种拆分方式和n(n-1)(n-2)/2种新的连接方式;或者,
19、③对目标蛋白质原有的三级结构中的m个环区进行拆分,共有n!/[(n-m)!×m!]种拆分方式和连接方式:种新的连接方式,其中m为4或大于4的整数,其中l为1到m-1的正整数;
20、在一些优选的实施方式中,按照需要重新连接的环区的数量由少到多的顺序依次进行后续的评价和设计。
21、根据本发明在第一方面提供的拓扑蛋白质的设计方法,步骤ii)中所述评价的依据为:
22、对于每个待连接的位置设定两个评价标准,即(a)待连接结构基元间的直线距离,和(b)待连接二级结构基元所能生成环区符合天然蛋白环区统计规律的概率;
23、综合上述评价标准(a)和(b),计算得到待连接位置生成新环区的概率;
24、考虑当前连接方式中所有需要重新生成的环区,计算整体生成概率,以所述概率作为依据对所有连接方式进行打分并排序,选取优势得分组依次进行设计。
25、在具体实施方式中,所述打分的基本原则为:
26、(a1)对蛋白质数据库(pdb)中所有环区的直线距离进行统计,计算每种直线距离对应的环区数目与总环区数目的比值,将该比值作为该直线距离下可生成环区的概率p1;
27、(b1)对pdb中所有的蛋白质环区间的直线距离和环区长度进行统计,得到特定直线距离下环区长度的概率分布,以最小溶剂可及路径在目标位置生成虚拟环区作为实际可生成环区的最小长度,以此长度为积分下限,以统计所得的当前直线距离下可采取的最长环区长度为积分上限,对上述所得概率分布进行积分计算,得到实际生成环区符合统计规律的概率p2;
28、(c1)以(a1)和(b1)计算的概率乘积作为当前位置实际可生成环区的概率p,即p=p1×p2;
29、(d1)将当前连接方式下所有待连接的位置计算得到的可生成环区的概率的乘积作为该连接方式的概率ptotal,即ptotal=∏pi,其中所述i为所有需要生成的新的环区数目,以此进行打分并排序,根据排序确定在后续拓扑结构设计中的优先级。
30、根据本发明在第一方面提供的拓扑蛋白质的设计方法,步骤iii)中按照特定的连接方式,在二级结构基元之间通过最小溶剂可及路径生成新的虚拟环区;每种连接方式下生成多于一条的虚拟环区,通过遍历所有虚拟环区的生成顺序的组合,来遍历新生成的虚拟环区之间所有可能的空间关系,设其总数为n。
31、通过高斯连接数或者纽结不变量的计算程序对每一种生成顺序下新生成的虚拟环区对应的目标蛋白质的拓扑结构进行判断,得到其中符合目标拓扑结构的数目m;
32、根据上述n和m计算当前连接方式下生成目标拓扑结构的概率为m/n,输出此概率作为当前连接关系下该目标拓扑结构的形成概率;
33、根据能够形成目标拓扑结构的连接关系和空间关系,确定实际环区的长度范围(例如,以相应生成的虚拟环区长度,即虚拟点数目l为其下限,以l+30为其上限),并根据其相对空间关系施加长度限制,对于其中相邻且彼此交叉的环区,为维持相对空间关系不变,要求相对远离疏水核心的环区长度与相对靠近疏水核心的环区长度这两者之差不小于其下限的长度差,以此界定各个环区的长度范围,从而输出符合目标拓扑结构的设计结果及其中对应的新生成的实际环区的长度范围。
34、根据本发明在第一方面提供的拓扑蛋白质的设计方法,步骤iv)中根据iii)中确定的实际环区的长度范围,从中选择更加符合步骤ii)中打分评价的依据的环区长度组合,作为各个实际环区中氨基酸的具体数目,并据此设计实际环区的氨基酸序列,
35、优选的,选择如下三种方法中的任一种设计实际环区的氨基酸序列:(a)直接设计具有目标氨基酸数目的柔性连接环区,该柔性连接环区包含酶切位点、亲和纯化标签、偶联反应后残留基元、原环区的部分或全部序列、或者由甘氨酸g和丝氨酸s组成的连接序列中的任一种或其任意组合;(b)通过相似结构基元搜索算法在pdb中搜索与待连接基元两个端点类似的结构,选取其中环区长度符合要求的作为环区序列;(c)通过计算机辅助的手段,设计具有目标长度的连接环区序列。
36、更优选的,所述酶切位点为烟草蚀纹病毒蛋白酶酶切位点(enlyfqg)、烟草叶脉斑点病毒蛋白酶酶切位点(etvrfqg)、肠激酶酶切位点(ddddk)、凝血因子xa蛋白酶酶切位点(idgr)或者welqut蛋白酶酶切位点(welq)中的任一种;所述亲和纯化标签为histag标签(hhhhhh)、strep-tag ii标签(wshpqfek)或者flag标签(dykddddk)中的任一种;所述偶联反应后的残留氨基酸序列为cfn、esgsgk、lpetg或nhv中的任一种;所述相似结构基元搜索算法为master、fragbag,或topofit中的任一种;所述计算辅助手段为rosetta loopmodelling、scuba、或foldx loopreconstruction方法中的任一种。
37、根据本发明在第一方面提供的拓扑蛋白质的设计方法,所述拓扑蛋白质的化学拓扑结构选自支化(branched)、多环(multicyclic)、纽结(knot)和链环(link)结构中的任一种及其任意组合;优选的,所述拓扑蛋白质为具有2个或更多个机械互锁环状结构的蛋白质索烃,或者为具有三叶草结、41结、51结或52结结构的纽结蛋白。
38、本发明在第二方面提供了一种拓扑蛋白质,其特征在于,所述拓扑蛋白质是根据本发明在第一方面提供的方法设计得到的拓扑蛋白质。
39、在一些具体的实施方式中,所述拓扑蛋白质的化学拓扑结构选自支化(branched)、多环(multicyclic)、纽结(knot)和链环(link)结构中的任一种及其任意组合;优选的,所述拓扑蛋白质为具有2个或更多个机械互锁环状结构的蛋白质索烃,或者为具有三叶草结、41结、51结或52结结构的纽结蛋白。
40、下文对本发明第一和第二方面提供的技术方案作进一步的解释和说明。
41、蛋白质的三级结构由二级结构基元(如α螺旋和β折叠)及连接二级结构基元的柔性环区共同组成,具有相对保守的结构,其中环区相对暴露,可工程化程度更高。通过改造环区对结构基元进行重新接线可在不大幅改变疏水核心的前提下实现蛋白质的化学拓扑改造。本发明在保留目标蛋白结构基元基本不变的前提下,通过计算机辅助的手段,设计出各种新的环区重新接线方式,从而将目标蛋白改造为多种具有特定化学拓扑结构的变体。
42、以下对本发明的技术方案进行详细说明。
43、本发明提供的拓扑蛋白质的程序化设计方法的整个设计流程包括以下四个主要步骤,并且可以实现完全的程序化。
44、1)对目标蛋白原有的三级结构进行合理拆分,根据目标蛋白质的化学拓扑结构,设计二级结构基元间的所有可能的重新连接方式
45、在三维空间中对目标蛋白质的二级结构基元进行重新连接存在数目众多的可能性。由于细胞合成的蛋白质前驱体均为线型结构,在此基础上,考虑n端和c端的虚拟连接在内,整个蛋白质可以被分为n个二级结构基元和n个环区。将所述二级结构基元重新连接得到单环纽结(包括平凡纽结的环状分子)的可能连接方式为(n-1)!(即n-1的阶乘)种,将所述二级结构单元重新连接得到多环链环的可能连接方式更多,如实现二元链环的可能连接方式为其中l为1到n-1的正整数。这些连接方式还可能因为环区和结构基元间的相对空间关系不同,而产生不同的化学拓扑结构。结合每个环区的序列设计,最终可能形成的蛋白质序列和构建更是不可胜数,而真正符合目标拓扑结构要求的连接方式仅占据其中很小的一部分。如果进一步考虑改变连接方式可能带来的结构不稳定性、折叠动力学壁垒、以及后续实际合成和制备中可能存在的问题(如合成过程中组装和反应不能很好协同,存在多种副反应等),可行性相对较高的拓扑构建可能只占据其中非常小的一部分。
46、因此在本发明中,不再遍历所有可能的拆分和连接方式,而是根据目标蛋白质的化学拓扑结构选择尽量少的环区进行工程化。
47、如图1中所示,以设计蛋白质[2]索烃(即包含2个环的蛋白质索烃,又可称为二元链环)为例,可仅选择所述蛋白质原有结构中的2个环区进行拆分,将得到的两部分多肽链的n端和c端分别环化,即可得到二元的链环结构。理论上,共有n(n-1)/2种拆分方式,每种拆分有且仅有一种新的连接方式,故最终共有n(n-1)/2种不同的新的连接方式。如果对所述蛋白质原有结构中的3个环区进行拆分,共有n(n-1)(n-2)/6种拆分方式,每种拆分后重新连接形成二元链环存在三种方式,因此共有n(n-1)(n-2)/2种连接形式,以此类推。从这些连接方式中,通过进一步设计空间关系,就有可能得到蛋白质[2]索烃结构。当然,还可以根据需要选择更多数目的环区进行拆分。例如,如果对所述蛋白质原有结构中的m个环区进行拆分,共有n!/[(n-m)!×m!]种拆分方式和种连接方式,其中m为4或大于4的整数,l为1到m-1的正整数。
48、由于工程化的环区数目越少,对折叠结构的扰动也就越小,最终成功合成拓扑蛋白质的概率也相对越高。按照环区重新接线数目由少到多的顺序依次设计,可保证优先设计成功率高的体系。
49、2)对二级结构基元间可能的重新连接方式进行打分评价,确定优先级
50、通过合理优化环区的拆分方式大大减少了可能的重新连接方式,随后针对每种连接方式,进行打分评价,选择得分高的体系优先进行下一步设计,其他得分低的体系次之。打分评价的依据包括:二级结构基元待连接端点的距离和所能生成环区符合天然蛋白环区统计规律的概率。
51、如图2中的(a)所示,对pdb(protein data bank,蛋白质数据库)中所有环区的直线距离进行统计,计算每种直线距离对应的环区数目与总环区数目的比值,将该比值作为该直线距离下可生成环区的概率p1。
52、如图2中的(b)所示,对pdb中所有的蛋白质环区间的直线距离和环区长度进行统计,得到特定直线距离下环区长度的概率分布,在设计过程中通过程序以最小溶剂可及路径(其中所述“溶剂可及路径”是指在蛋白质表面两点间生成的不与蛋白质发生碰撞的路径,而其中最短的路径即为“最小溶剂可及路径”,此时生成的路径与蛋白质表面间无法容纳另一条路径穿过,具体参见bioinformatics,2019,35,3169-3170)在目标位置生成虚拟环区作为实际可生成环区的最小长度,以此长度为积分下限,以统计所得的当前直线距离下可采取的最长环区长度为积分上限,对上述所得概率分布进行积分计算,可得实际生成环区符合统计规律的概率p2。具体计算溶剂可及距离的算法如下:以待连接氨基酸中点为中心,将周围半径为5nm的空间进行网格划分,每个网格大小为0.1nm,在网格中未被蛋白质占据的空间进行随机行走,遍历所有可能的连接两个氨基酸路径,并从中选择最短路径作为最小溶剂可及路径。
53、将基于上述两种影响因素计算的概率(即上文所述概率p1和概率p2)乘积作为当前位置实际可生成环区的概率p(即p=p1×p2)。
54、最后将当前连接方式下所有待连接位置计算得到的可生成环区的概率的乘积作为该连接方式的概率ptotal,ptotal=∏pi,其中所述i为所有需要生成的新的环区数目,以此进行打分并排序,根据排序确定在后续拓扑结构设计中的优先级。
55、3)针对每种连接方式,依次生成新的虚拟环区,遍历所有的生成顺序组合,产生相应的空间关系,判断连接后的化学拓扑结构,计算得到该连接方式下目标拓扑结构的形成概率,并确定新生成的环区长度范围。
56、在确定了所有可能的拆分方式和连接方式后,根据步骤2)中的打分排序,即可对每种可能的连接方式进行下一步设计。
57、下面仍以蛋白质[2]索烃结构的设计为例进行说明。
58、如图3所示,对蛋白中的每个二级结构基元进行编号(例如可以将所述蛋白中的6个二级结构基元顺序编号为a、b、c、d、e和f),相应的环区以其连接的两个二级结构基元的编号进行表示,如连接二级结构基元a和二级结构基元b的环区为环区ab,连接目标蛋白质原有结构中的n端和c端的环区为环区fa。对其中一对环区(具体为环区fa/环区de)进行了拆分,为形成索烃的结构,可能采取的连接方式只有一种,即重新生成环区da和环区fe。随后分别生成虚拟环区,并遍历所有的生成顺序组合。
59、虚拟环区通过计算最小溶剂可及路径的算法进行生成。其中所述“溶剂可及路径”的定义同上文。以此最小溶剂可及路径作为虚拟环区,可以保证先生成的虚拟环区紧贴蛋白质表面,而后续生成的环区位于蛋白质表面和先生成环区的同一侧,这样环区的生成顺序就决定了环区的相对空间位置。通过遍历虚拟环区所有的生成顺序组合就可以遍历所有环区的相对空间位置,假设其总数为n(即所有的生成顺序组合)。随后,对每种空间位置组合得到的蛋白质的拓扑结构进行判断,判断是通过高斯连接数或者纽结不变量的计算程序执行的。统计其中可以形成蛋白质[2]索烃的数目为m,以m/n作为当前连接方式下索烃结构的形成概率。同时该设计过程也给出具有目标化学拓扑结构的蛋白质改造方式,及相应新生成的虚拟环区的长度范围。
60、本发明提供的程序化设计方法通过优化体系的拆分方式、对所有可能的连接方式进行打分评价等手段大大减少了整个设计过程中需要遍历的可能性数目,提高了设计拓扑蛋白质的效率。
61、4)设计拓扑蛋白质新生成的环区序列
62、根据步骤3)计算得到的虚拟环区的长度范围,从中优选更加符合步骤2)中打分标准的环区长度组合,作为各个实际环区中氨基酸的具体数目,并据此设计实际环区的氨基酸序列。
63、具体可以采用如下三类设计方法中的任一种设计实际环区的氨基酸序列:(a)直接设计目标氨基酸数目的柔性连接环区,该环区包含酶切位点、亲和纯化标签、偶联反应后残留基元、原环区的部分或全部序列或者由甘氨酸g和丝氨酸s组成的连接序列中的任一种或其任意组合;(b)通过master(protein sci.2015,24,508-524)、fragbag(pnas 2010,107,3481-3486)或topofit(protein sci.2004,13,1865-1874)中的任一种相似结构基元搜索算法在pdb中搜索与待连接的二级结构基元两个端点类似的结构,选取其中环区长度符合要求的作为目标环区的序列;或者,(c)通过计算机辅助手段,如rosetta loopmodelling(nat.method 2009,6,551-552)、scuba(nature 2022,602,523-528)或foldxloopreconstruction(plos comput.biol.2008,4,p.e1000083)方法中的任一种设计具有目标长度的连接环区序列。
64、发明定义
65、本文中所用术语的选择,旨在最好地解释各实施例的原理、实际应用或对市场中的技术的改进,或者使本技术领域的其它普通技术人员能理解本文披露的各实施例。除非另有定义,否则本文中使用的所有技术和科学术语均具有与本领域一般技术人员通常所理解的含义相同的含义。为了本发明的目的,下文定义了以下术语。
66、术语“约”在与数字数值联合使用时意为涵盖具有比指定数字数值小5%的下限和比指定数字数值大于5%的上限的范围内的数字数值。
67、术语“和/或”当用于连接两个或多个可选项时,应理解为意指可选项中的任一项或可选项中的任意两项或多项。
68、如本文中所用,术语“包括”意指包括所述的要素、整数或步骤,但是不排除任意其他要素、整数或步骤。在本文中,当使用术语“包括”时,除非另有指明,否则也涵盖由所述及的要素、整数或步骤组成的情形。
69、术语“数值a~数值b”表示的数值范围是指包含端点数值a、b的范围。
70、术语“以上”或“以下”表示的数值范围是指包含本数的数值范围。
71、术语“可以”表示的含义包括了进行某种处理以及不进行某种处理两方面的含义。
72、术语“任选”或“任选的”表示某些物质、组分、执行步骤、施加条件等因素使用或者不使用。
73、术语“蛋白质[2]索烃”表示包含2个环的蛋白质索烃,又可称为蛋白质二元链环。
74、术语“三叶草结、41结、51结、52结”表示纽结理论中标准命名法定义的三种不同纽结结构。其中三叶草结表示在二维平面上投影后含有的最小交叉点数为3的纽结;41结表示在二维平面上投影后含有的最小交叉点数为4的纽结;51结表示在二维平面上投影后含有的最小交叉点数为5,且最大对称性为c5对称性的纽结;52结表示在二维平面上投影后含有的最小交叉点数为5,且最大对称性为c2对称性的纽结。
75、本说明书中,所提及的“一些具体/优选的实施方案”、“另一些具体/优选的实施方案”、“实施方案”等是指所描述的与该实施方案有关的特定要素(例如,特征、结构、性质和/或特性)包括在此处所述的至少一种实施方案中,并且可存在于其它实施方案中或者可不存在于其它实施方案中。另外,应理解,所述要素可以任何合适的方式组合在各种实施方案中。
76、发明的效果
77、由本发明的技术方案可见,本发明的技术方案与现有技术相比,具有以下有益效果:
78、(1)本发明提供的拓扑蛋白质的设计方法为阐明拓扑结构在蛋白质构效关系中的作用提供理想的平台,也为开发拓扑功能蛋白质提供了便捷的方法。
79、(2)本发明提供了一种对于多种蛋白质广泛适用的拓扑异构体设计策略,整个策略流程清晰,可操作性强且可以完全实现程序化。结合合适的环化策略可以指导蛋白质拓扑结构的有效合成。
80、为了让本发明的上述和其他目的、特征和优点能更明显易懂,下面特举较佳实施例,并配合说明书附图,作详细说明如下:
本文地址:https://www.jishuxx.com/zhuanli/20240615/86229.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表