一种低表达ACTB蛋白的肿瘤细胞系模型的制作方法
- 国知局
- 2024-06-20 11:20:02
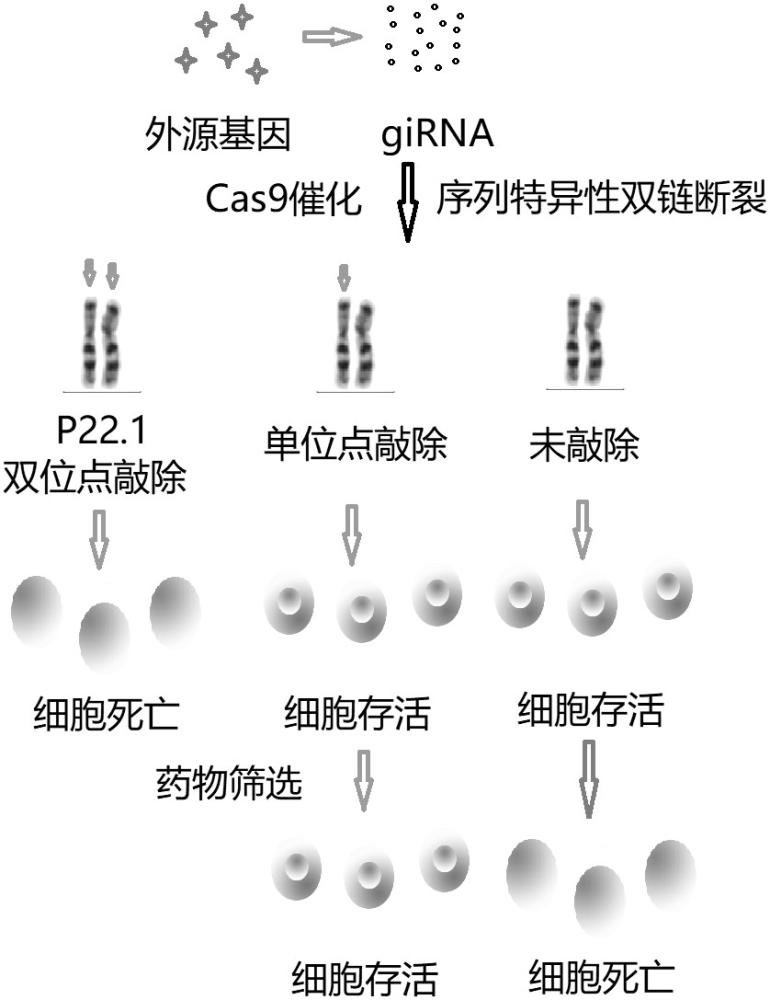
本发明属于肿瘤细胞模型,具体涉及一种低表达actb蛋白的肿瘤细胞系。
背景技术:
1、多发性肿瘤转移是肿瘤治疗失败的主要原因。肿瘤转移的主要途径有直接侵袭、腔室播散、经血循转移、经淋巴管转移等。肿瘤细胞在转移出原发病灶的时候需要变形,同原有的基质解离并突破基质的限制。肿瘤细胞转移到一个新的地方,又需要突破周边基质的限制并进入新的位点,形成新的细胞粘附点。在此过程中需要细胞变形、运动。actin即“肌动蛋白”,是细胞的一种重要骨架蛋白。actin在细胞分泌、吞噬、移动、胞质流动和胞质分离等过程中起重要作用。actin在不同物种之间高度保守,大致可分为六种,其中四种是不同肌肉组织特异性的,包括α-skeletalmuscleactin,α-cardiacmuscleactin,α-smoothmuscleactin,和γ-smoothmuscleactin;其余两种广泛分布于各种组织中,包括β-actin(β-non-muscle)和γ-non-muscleactin。β-actin是横纹肌肌纤维中的一种主要蛋白质成分,也是肌肉细丝及细胞骨架微丝的主要成分。具有收缩功能,分布广泛。β-actin(actb)即β肌动蛋白,在细胞中mrna表达数量很高,是肌肉细丝及细胞骨架微丝的主要成分。在维持细胞结构、细胞内运动、细胞分裂等细胞生理活动方面发挥着重要的作用,该基因几乎在所有真核细胞中表达,广泛存在哺乳动物动物的组织与细胞内。
2、肌动蛋白的组装和解聚主要受到rhogtpases信号通路的调节。其中,rhogtpases家族包括rhoa、rac1、cdc42等几个重要的成员。这些蛋白通过与相关的下游效应蛋白相互作用,调控不同的细胞过程。例如,rhoa可以促进肌动蛋白的聚集和稳定,促进细胞的收缩和增强细胞-细胞的黏附能力。而rac1和cdc42可以促进肌动蛋白的解聚和增加细胞外基质附着物(ecm)的聚集能力,从而加强细胞在环境中的黏附和迁移能力。一些辅助调节机制会影响肌动蛋白的组装和解聚。例如:磷酸化可以增强rhogtpases家族成员的活性,促进细胞的收缩和黏附能力。而去磷酸化则可以加强rac1和cdc42的作用,促进肌动蛋白的解聚,并增加细胞在环境中的移动能力。
3、越来越多的证据表明,actb在多种癌症中异常表达,actb的异常表达改变了细胞骨架,影响着肿瘤的侵袭和转移。actb的表达与肿瘤患者预后存在着显著的关联。在大多数癌症中,actb表达与免疫细胞浸润有关,同时与免疫检查点和其他免疫调节剂相关。此外,粘着斑和肌动蛋白调节相关途径也受actb表达的影响。actb可以通过nf-κb和wnt/β-catenin通路抑制头颈部鳞状细胞癌细胞迁移和侵袭。多数专家认为,actb可能是癌症中预后不良和免疫浸润的潜在生物标志物。
4、为了研究肿瘤细胞的迁移、侵袭规律,研究中常常需要低actb表达细胞。研究抑制肿瘤迁移的药物也需要actb低表达细胞。这个细胞系将成为众多医学和生物学研究实验室的重要细胞系。预估市场量极大。因此,我们计划制备这一细胞系。然而actb是细胞必须基因之一。敲除actb将会导致细胞死亡。因此我们采用压力选择技术,结合基因敲除技术制成一株稳定低表达actb基因的细胞,以满足生产要求。为此我们发明了一种低表达actb蛋白的肿瘤细胞系来解决上述问题。
技术实现思路
1、本发明的目的是提供一种低表达actb蛋白的肿瘤细胞系,能够表达低水平actb,适用于肿瘤研究中的多个方面。通过使用杂合子技术降低重要基因表达,但是无损细胞增殖能力,经基因和蛋白水平验证,actb基因表达大幅下降。相对于瞬时转染,本细胞系具有结果稳定,成本低廉,简单传代后可满足用户需求的优势。
2、本发明采取的技术方案具体如下:
3、一种低表达actb蛋白的肿瘤细胞系,2023年11月29日保藏于中国微生物菌种保藏管理委员会普通微生物中心,科学描述为:actb敲低293t细胞,保藏号为cgmcc no.45753,所述肿瘤细胞系的制备方法包括以下步骤:
4、在一种优选方案中,所述材料为仪器、耗材、试剂及其他物品,所述仪器包括净化工作台、离心机、恒温水浴箱、冰箱、倒置显微镜、培养箱、微量加样器,所述耗材包括枪头、培养皿、15ml离心管、酒精灯、废液缸,所述试剂包括培养基、血清、双抗、胰蛋白酶、1×pbs,所述双抗为青霉素及链霉素,所述其他物品为记号笔、记号笔、橡胶手套、口罩、100%和75%酒精。基因合成服务和关键质粒均购自上海碧云天生物技术有限公司。
5、在一种优选方案中,所述细胞的复苏为从液氮中取出冻存管,直接浸入37℃温水中,摇动使其快速融化。从水浴中取出冻存管,在超净台中打开盖子,用枪头吸出细胞悬液,加到已预先加入了3ml细胞完全培养基的15ml离心管中,轻弹混匀。1000rpm,离心5min。弃去上清液,加入含10%fbs的细胞培养基,重悬细胞。调整细胞密度,接种培养皿。37℃培养箱静置培养,次日更换培养液。
6、在一种优选方案中,所述细胞的传代为将长成单层的细胞培养皿从二氧化碳培养箱中取出。在超净工作台中,吸出瓶内的培养基,加入1×pbs2ml,轻轻旋转培养皿以清洗细胞。吸弃pbs,加入胰蛋白酶0.5ml,静置3-5分钟。待细胞变圆,相互之间不再连接成片时,加入2倍体积完全培养基,吹打,制成细胞悬液。将细胞悬液吸出置于15ml离心管,1000rpm,离心5min,弃上清液。加入1.5ml完全培养基,吹打,混匀细胞。取出另外两个新的60mm培养皿,每个加入2.5ml完全培养基。原消化皿中也加入2.5ml完全培养基,将离心管中的细胞悬液0.5ml/皿,梅花式分别滴入三个培养皿中,用枪头吹打细胞若干次,置于二氧化碳培养箱中培养。
7、在一种优选方案中,所述细胞转染为胰酶消化细胞并计数,接种至100mm培养皿中,使其在转染日密度为60%-70%。底面积为100mm的细胞培养皿,每个皿内加入14.0μg质粒dna,用1.5ml无血清培养基稀释,混合在室温下放置5min。使用1.5ml无血清培养基稀释80μllipofectaminie2000试剂。同稀释的dna混合,室温静置20分钟。将上述混合物均匀的加入到细胞中,在37℃,5%co2,100%饱和湿度中保温6小时。每个培养皿各加入12ml含10%fbs的dmem培养液,继续培养。24小时后用含10%fbs的dmem培养液替换原先的旧培养基。转染48小时后加入新霉素至终浓度200ug/ml,继续培养48小时,幸存细胞挑单克隆,继续培养。
8、在一种优选方案中,所述actb片段扩增为从培养箱取出细胞,吸取培养瓶内液体,使用常温pbs清洗一遍,吸弃pbs,使用细胞刮刀刮下细胞,用移液器将刮下的细胞悬液转移至1.5ml离心管,反复吹打30~60次,样品离心管插于冰上。配置pcr反应体系,10×扩增缓冲液10ul,4种dntp混合物各200umol/l,引物各10~100pmol,模板dna0.1~2ug,taqdna聚合酶2.5u,mg2+1.5mmol/l,加双蒸水至100ul,进行基因扩增反应,dna预变性:96℃3min,后续dna预变性:96℃30s,退火:55℃30s,延伸:72℃60s,72℃300s,以上条件做30组扩增循环。
9、在一种优选方案中,所述琼脂糖凝胶电泳为称取琼脂糖溶解在电泳缓冲液中,按0.3-1.5%的琼脂糖含量,用1%的凝胶,置微波炉中加热至完全溶化,摇匀,将冷却到60℃的琼脂糖溶液轻轻倒入电泳槽水平板上,待琼脂糖胶凝固后,在电泳槽内加入电泳缓冲液,拔出梳子,将dna样品与加样缓冲液按4:1混匀后,用微量移液器将混合液加到样品槽中。每槽加10-20μl,电压3-5v/cm,电泳0.5hr,当溴酚蓝移到距凝胶前沿1-2cm时,停止电泳。取出凝胶,放在含有溴化乙锭的染色液中染色30min,在254nm的紫外灯下观察,照相记录电泳图谱。
10、在一种优选方案中,所述测序鉴定为贴壁细胞用胰酶消化,离心收集,细胞重悬于冰冷的pbs漂洗一次,离心收集,重复两次,加入5mldna提取缓冲液,10mmol/ltris-cl,0.1mol/ledta,0.5%sds,混匀,加入25ul蛋白酶k,使终浓度达到100ug/ml,混匀,50℃水浴3h,用等体积的酚抽提一次,2500r/min离心收集水相,用等体积的酚,氯仿,异戊醇混合物抽提一次,2500r/min离心收集水相,用等体积的氯仿,异戊醇抽提一次,加入等体积的5mol/l的licl,混匀,冰浴,10min,2500r/min,离心10min,转上清与一离心管中,加入等体积的异丙醇,室温10分钟,2500r/min,离心10min,弃上清,加入0.1倍体积3mol/l乙酸钠,ph5.2,与2倍体积-20℃预冷无水乙醇,-20℃20分钟,12000r/min,室温离心5分钟,弃上清,将dna溶于适量te中,测序并比较序列。
11、本发明取得的技术效果为:
12、制备了一个低表达actb的肿瘤细胞系,可广泛用于肿瘤迁移、侵袭、粘附、转移、耐药、增殖等多领域研究,有广泛的学术或者商业价值使用杂合子技术,降低了生命重要基因的表达,但是不影响细胞的分裂增殖;
13、改变了细胞系基因组,即可获得基因的稳定改变,本细胞系经基因水平,蛋白水平双重验证,结果稳定可靠;
14、本细胞系筛选成型后,只需简单传代即可满足生产需求,成本低廉。
本文地址:https://www.jishuxx.com/zhuanli/20240619/1401.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表