一种1,2,3-三氮唑-4-甲酸乙酯的合成方法与流程
- 国知局
- 2024-09-14 14:31:44
本发明属于有机化学合成,具体涉及一种1,2,3-三氮唑-4-甲酸乙酯的合成方法。
背景技术:
1、1,2,3-三氮唑-4-羧酸酯类化合物是一种重要的药物合成砌块,广泛应用于连接各种功能分子片段。在药物化学研究中,1,2,3-三氮唑除了作为连接链出现在各种活性分子当中,还有一个重要角色,那就是可以作为生物电子等排体。已被用于内脏利什曼病的临床前候选药物dndi-6148(journal of medicinal chemistry,2021,vol.64,#21,p.16159-16176),smhdac8抑制剂(chemmedchem,2020,vol.15,#7,p.571-584),抗丝分裂剂(bioorganic chemistry,2019,vol.83,p.535-548),ccr2受体拮抗剂(jp5431316),蛋白质/肽修饰分子(us2022/204481),钠通道药理学调节剂(us2019/60284),抗癫痫药物鲁非那胺(tetrahedron letters,2010,vol.51,#24,p.3229-3231),认知障碍苯二氮卓衍生物(jp2020/512389)等等的合成,但目前还没有可用于中试放大的工艺路线。
2、已有的合成1,2,3-三氮唑-4-甲酸乙酯的工艺报道较少。文献(journal ofheterocyclic chemistry,1989,vol.26,p.1461-1468)中报道了该化合物的合成方法,采用叠氮苯与丙炔酸乙酯为原料,五水硫酸铜,抗坏血酸钠和(e)-n-(1-{[(1-苯甲基-1h-1,2,3-三唑-4-基)甲基][(2e)-2-(肟基)丙基]氨基}丙烷-2-亚基)羟胺为反应试剂,在甲醇、水中反应(见下图),柱分离得到了最终产物。原料叠氮苯系易爆品,丙炔酸乙酯为催泪剂,无论安全性还是后处理操作均不适合中试生产。
3、
4、专利us2010/160323也报道了1,2,3-三氮唑-4-甲酸乙酯的合成方法(见下图),采用乙基2-重氮基-3-氧亚基丙酯和苯胺在乙酸/乙醇中反应12小时,反应液浓缩后经中水析出,得到最终产物。该路线所用原料乙基2-重氮基-3-氧亚基丙酯没有市场供应,须现做现用,且极不稳定。因此也不适合作为中试工艺路线。
5、
技术实现思路
1、本发明要解决的技术问题在于提供一种合成1,2,3-三氮唑-4-甲酸乙酯的合成方法,原料和试剂都较稳定、经济易得,反应副产物少、收率高,后处理简单,易于放大生产,成本较低。
2、为解决上述技术问题,本发明采用的具体技术方案如下:
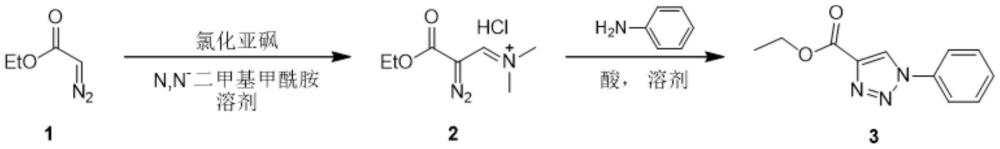
3、本发明提供一种1,2,3-三氮唑-4-甲酸乙酯的合成方法,步骤一,以重氮乙酸乙酯(化合物1)作原料,在氯化亚砜、n,n-二甲基甲酰胺、溶剂存在下发生反应生成中间体(2-重氮基-3-乙氧基-3-氧亚基亚丙基)盐酸二甲基氮烷正离子(化合物2);步骤二,中间体在苯胺、酸、溶剂存在下发生反应生成1,2,3-三氮唑-4-甲酸乙酯;
4、
5、进一步的,一种1,2,3-三氮唑-4-甲酸乙酯的合成方法,包括如下步骤:
6、步骤一:氯化亚砜加入到n,n-二甲基甲酰胺中,经加热、反应、浓缩得到固体;加入溶剂,加入重氮乙酸乙酯,反应后得到中间体化合物2;
7、步骤二:将中间体化合物2溶于溶剂中,加入苯胺、酸,反应后得到产物。
8、在一些具体实施方案中,所述步骤一中的溶剂选自氯仿、甲苯、四氢呋喃、乙酸乙酯、二氯甲烷中的一种或多种。
9、在一些具体实施方案中,所述步骤二中的溶剂为水与有机溶剂的混合物,所述有机溶剂选自1,4-二氧六环、乙醇、四氢呋喃、甲醇、n,n二甲基甲酰胺、二甲基亚砜中的一种或多种。
10、在一些具体实施方案中,所述步骤一,氯化亚砜加入到n,n-二甲基甲酰胺中后的加热温度为30~80℃,反应时间为0.5~15小时;优选为加热温度为40~50℃,反应时间为2~3小时。
11、在一些具体实施方案中,所述步骤一,控制反应体系温度为0~20℃时加入重氮乙酸乙酯,加入方式为滴加,加入时间为0.5~1小时。
12、在一些具体实施方案中,所述步骤一,完成重氮乙酸乙酯的加入后,在0~50℃下,反应1~18小时;优选为在20~30℃下,反应12~15小时。
13、在一些具体实施方案中,所述步骤二,反应温度为0~80℃,反应时间为2~24小时;优选为反应温度为20~30℃,反应时间为12~15小时。
14、在一些具体实施方案中,所述步骤一还包括后处理,具体步骤为将所述步骤一中得到的固体在n2气氛下过滤,用甲基叔丁基醚(mtbe)打浆,过滤收滤饼,得到中间体化合物2;所述步骤二还包括后处理,具体步骤为反应后加水稀释,提取后合并有机相,经洗涤、干燥、过滤、浓缩后,打浆,过滤后浓缩滤饼得到产物。
15、在一些具体实施方案中,所述步骤二中的重结晶溶剂为异丙醚、甲基叔丁醚、正己烷、石油醚、乙酸乙酯的单一品种或混合物,温度为0~50℃,优选为25℃。
16、在一些具体实施方案中,所述步骤一中,重氮乙酸乙酯、n,n-二甲基甲酰胺、氯化亚砜的投料比为1~4:0.5~2:0.5~2;所述步骤二中,(2-重氮基-3-乙氧基-3-氧亚基亚丙基)盐酸二甲基氮烷正离子、苯胺、醋酸的投料比为1~4:1~4:0.25~2。
17、与现有技术相比,本发明具备如下有益效果:本发明采用的方法避免了叠氮底物的使用,安全性更高,且工艺原料和试剂都是稳定、经济易得的,反应副产物少、收率高,两步反应后处理简单,适合中试放大,是一种合成1,2,3-三氮唑-4-甲酸乙酯的经济可行的工艺。
18、以下将对本发明的构思、具体结构及产生的技术效果作进一步说明,以充分地了解本发明的目的、特征和效果。
技术特征:1.一种1,2,3-三氮唑-4-甲酸乙酯的合成方法,其特征在于,步骤一,以重氮乙酸乙酯作原料,在氯化亚砜、n,n-二甲基甲酰胺、溶剂存在下发生反应生成中间体化合物2(2-重氮基-3-乙氧基-3-氧亚基亚丙基)盐酸二甲基氮烷正离子;步骤二,中间体在苯胺、酸、溶剂存在下发生反应生成1,2,3-三氮唑-4-甲酸乙酯;
2.根据权利要求1所述的合成方法,其特征在于,包括如下步骤:
3.根据权利要求1或2任一项所述的合成方法,其特征在于,所述步骤一中的溶剂选自氯仿、甲苯、四氢呋喃、乙酸乙酯、二氯甲烷中的一种或多种。
4.根据权利要求1或2任一项所述的合成方法,其特征在于,所述步骤二中的溶剂为水与有机溶剂的混合物,所述有机溶剂选自1,4-二氧六环、乙醇、四氢呋喃、甲醇、n,n二甲基甲酰胺、二甲基亚砜中的一种或多种。
5.根据权利要求1或2任一项所述的合成方法,其特征在于,所述步骤一,氯化亚砜加入到n,n-二甲基甲酰胺中后的加热温度为30~80℃,反应时间为0.5~15小时。
6.根据权利要求1或2任一项所述的合成方法,其特征在于,所述步骤一,控制反应体系温度为0~20℃时加入重氮乙酸乙酯,加入方式为滴加,加入时间为0.5~1小时。
7.根据权利要求1或2任一项所述的合成方法,其特征在于,所述步骤一,完成重氮乙酸乙酯的加入后,在0~50℃下,反应1~18小时。
8.根据权利要求1或2任一项所述的合成方法,其特征在于,所述步骤二,反应温度为0~80℃,反应时间为2~24小时。
9.根据权利要求1或2任一项所述的合成方法,其特征在于,所述步骤一还包括后处理,具体步骤为将所述步骤一中得到的固体在n2气氛下过滤,用甲基叔丁基醚(mtbe)打浆,过滤收滤饼,得到中间体化合物2;所述步骤二还包括后处理,具体步骤为反应后加水稀释,提取后合并有机相,经洗涤、干燥、过滤、浓缩后,打浆,过滤后浓缩滤饼得到产物。
10.根据权利要求1或2任一项所述的合成方法,其特征在于,所述步骤一中,重氮乙酸乙酯、n,n-二甲基甲酰胺、氯化亚砜的投料比为1~4:0.5~2:0.5~2;所述步骤二中,(2-重氮基-3-乙氧基-3-氧亚基亚丙基)盐酸二甲基氮烷正离子、苯胺、醋酸的投料比为1~4:1~4:0.25~2。
技术总结本发明公开了一种1,2,3‑三氮唑‑4‑甲酸乙酯的合成方法,步骤一,以重氮乙酸乙酯作原料,在氯化亚砜、N,N‑二甲基甲酰胺、溶剂存在下发生反应生成中间体化合物2(2‑重氮基‑3‑乙氧基‑3‑氧亚基亚丙基)盐酸二甲基氮烷正离子;步骤二,中间体在苯胺、酸、溶剂存在下发生反应生成1,2,3‑三氮唑‑4‑甲酸乙酯。本发明采用的方法避免了现有技术中叠氮底物的使用,安全性更高,且工艺原料和试剂都是稳定、经济易得的,反应副产物少、收率高,两步反应后处理简单,适合中试放大,是一种合成1,2,3‑三氮唑‑4‑甲酸乙酯的经济可行的工艺。技术研发人员:吴潮波,杨念勇,杨生涛,陈俊一,高宇,徐艳受保护的技术使用者:南通药明康德医药科技有限公司技术研发日:技术公布日:2024/9/12本文地址:https://www.jishuxx.com/zhuanli/20240914/294578.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表