基于残差网络和序列特性的蛋白质分子识别特征预测方法
- 国知局
- 2024-10-21 14:36:30
本发明涉及生物信息学,特别是一种基于残差网络和序列特性的蛋白质分子识别特征预测方法。
背景技术:
1、内源性无序蛋白质(intrinsically disorderedproteins,idps)是一类在天然状态下缺乏恒定三维结构的蛋白质,在细胞信号传导和转录调控等生物过程发挥着关键作用。最新研究表明,内源性无序蛋白质在阿尔茨海默病、帕金森病及某些癌症等疾病的发生中也起着作用。分子识别特征区域(molecular recognition features,morfs)作为内源性无序蛋白质的一种关键功能区域,在与配体蛋白质结合时能够从无序状态转换为有序状态,其结构的高度灵活性使morfs区域与配体蛋白质能够精准结合,发挥着细胞信号传递和选择性剪接过程中的“分子开关”作用。研究表明大量具有识别功能的蛋白质集中在morfs区域,morfs区域的功能与多种疾病紧密相关,因此预测和理解morfs区域对于发现新的药物靶点具有重要生物医学价值。
2、近年来,许多morfs预测方法被提出,主要分为两类。一类是基于多特征融合的预测方法,例如,morfpred[1]利用五种不同类型的特征训练支持向量机(svm)进行预测;morfchibi[2]基于物化学性质训练两个独立的svm。另一类是组合预测,例如,morfchibi_light[3]利用bayes规则将espritz和morfchibi得到的分数进行合并;morfchibi_web[3]融合morf保护评分(mcs)、内在无序预测(idp)和morfchibi得分;opal[4]集成morfchibi和promis的预测因子进行预测。虽然这些方法明显提高了预测准确性,但是也增加了计算复杂度,并且在特征提取时只计算一次均值,忽略了特征信息的局部细化和细微差异。
技术实现思路
1、本发明旨在克服现有技术的局限性,提出一种基于残差网络和序列特性的蛋白质分子识别特征区域预测方法。
2、本发明解决所述技术问题采用如下的技术方案:
3、一种基于残差网络和序列特性的蛋白质分子识别特征预测方法,其特征在于,该方法包括如下步骤:
4、s1:对蛋白质序列特性进行筛选,得到四个特征集,前三个特征集分别包括多个序列特性,第四个特征集包括多个反映进化信息的序列特性;
5、s2:对待预测蛋白质序列进行预处理,提取待预测蛋白质序列各个残基关于每个特征集的特征矩阵;
6、首先在待预测蛋白质序列的两端分别添加(n-1)/2个零,得到扩展蛋白质序列,n为正整数;将扩展蛋白质序列针对特征集中的每个序列特性分别进行数字映射,得到各个序列特性对应的映射蛋白质序列;利用长度为n的滑动窗口对扩展蛋白质序列进行截取,生成多个残基片段;
7、根据序列特性对应的映射蛋白质序列,计算映射蛋白质序列中任意残基片段的平均映射值,将平均映射值作为残基片段的特征值并赋值给残基片段的各个残基;将残基的所有赋值进行累加求平均,得到残基的特征值;分别计算特征集中各个序列特性下残基的特征值,这些特征值组成残基的候选特征向量;对残基附近的多个残基的候选特征向量进行求和取平均,得到残基的特征向量,则第j个残基的特征向量xj表示为:
8、
9、式中,vm是第m个残基的候选特征向量,l是待预测蛋白质序列的长度;
10、针对每个特征集,分别选取短、中、长三种长度的滑动窗口对待预测蛋白质序列进行预处理,提取各个残基的特征向量,进而得到各个残基关于每个特征集的特征矩阵;
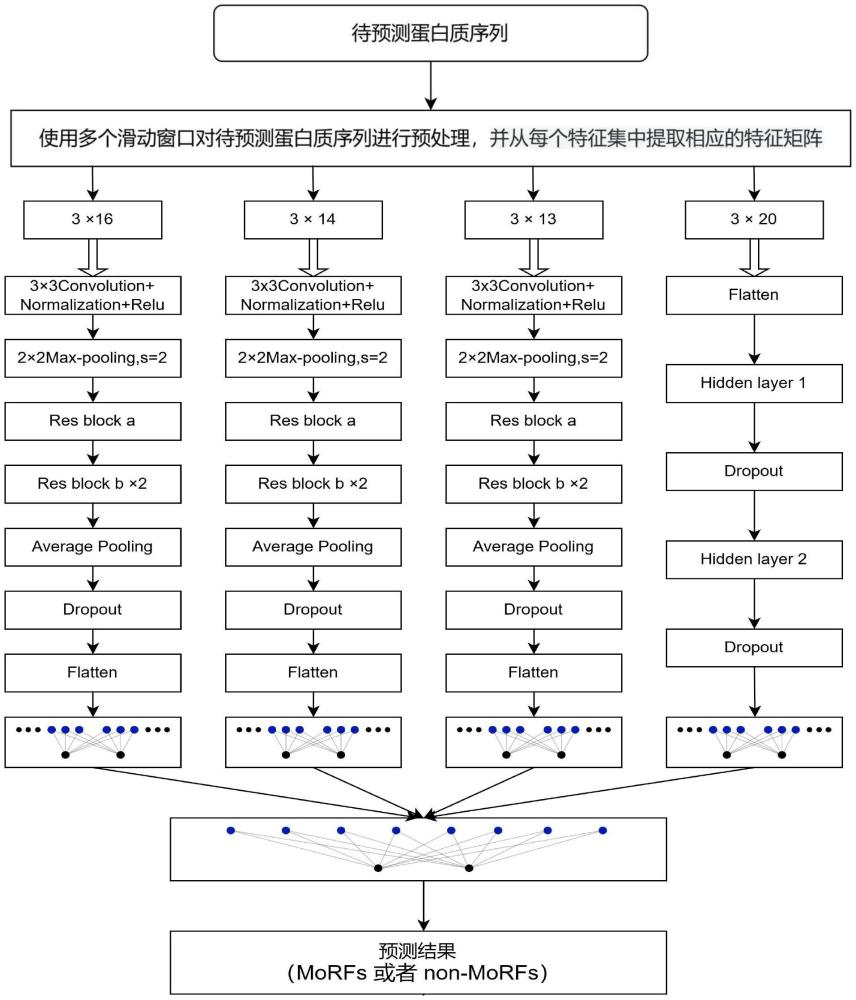
11、s3:构建预测模型;预测模型包含四个并行分支,四个分支的输入分别是残基关于四个特征集的特征矩阵,四个分支的输出经过全连接层,得到预测结果;
12、s4:对预测模型进行训练,将训练后的预测模型用于蛋白质分子识别特征区域的预测。
13、进一步的,所述预测模型的前三个分支的结构相同,输入的特征矩阵依次经过卷积、归一化和激活操作后,再经过最大池化操作,然后经过一个残差块a和两个残差块b,再经过丢弃层和展平操作,得到输出特征;对于第四个分支,输入的特征矩阵经过展平操作后,再依次经过第一隐藏层、第一丢弃层、第二隐藏层和第二丢弃层,得到第四个分支的输出特征。
14、进一步的,所述残差块a包含多个大小为3×3的卷积层,残差块a的输入特征与最后一个卷积层的输出特征进行跳跃连接,得到残差块a的输出特征;残差块b包含多个大小为3×3的卷积层,残差块b的输入特征经过1×1的卷积层后,再与最后个卷积层的输出特征进行跳跃连接,得到残差块b的输出特征。
15、进一步的,第一个特征集包含16个序列特性,其中13个是从氨基酸指数的多种氨基酸特性中筛选得到的主要影响内源性无序蛋白质分子识别特征区域的氨基酸特性,剩余3个是反映蛋白质序列的结构特性,一个是待预测蛋白质序列的拓扑熵,另外两个是待预测蛋白质序列的氨基酸倾向性;第二个特征集和第三个特征集分别包含14和13个从氨基酸指数的多种氨基酸特性中筛选得到的主要影响内源性无序蛋白质分子识别特征区域的氨基酸特性;第四个特征集包含20个反映进化信息的序列特征,基于ncbi非冗余数据库的蛋白质序列并根据氨基酸的位置特异性得分矩阵筛选得到。
16、与现有技术相比,本发明的有益效果是:
17、1.在待预测蛋白质序列预处理过程中,采用双均值计算策略,首先将残基片段的特征值赋予片段中的各个残基,随着滑动窗口的移动,每个残基被多次赋值,对所有赋值进行累加求平均,得到残基的特征值,所有序列特性下残基的特征值组成了残基的候选特征向量;然后,为了细化特征信息的局部变化和细微差异,将残基相邻的多个残基的候选特征向量进行求和取平均,得到最终的特征向量,充分提取了特征信息,提高了预测精度。
18、2.与常见的morfs预测方法不同,本发明采用单一预测器的形式进行预测,不需要多特征融合或者多种算法组合预测,大大简化了预测流程,降低了模型的计算复杂度,在提高了准确性的同时减少了计算资源的消耗,提升了运行效率和系统稳定性。仿真结果显示,本发明在保证较高tpr(真实阳性率)时,能够维持相对较低的fpr(假阳性率),显著减少了错误预测,提升了预测可靠性和精度,同时具有良好的鲁棒性。
技术特征:1.一种基于残差网络和序列特性的蛋白质分子识别特征预测方法,其特征在于,该方法包括如下步骤:
2.根据权利要求1所述的基于残差网络和序列特性的蛋白质分子识别特征预测方法,其特征在于,所述预测模型的前三个分支的结构相同,输入的特征矩阵依次经过卷积、归一化和激活操作后,再经过最大池化操作,然后经过一个残差块a和两个残差块b,再经过丢弃层和展平操作,得到输出特征;对于第四个分支,输入的特征矩阵经过展平操作后,再依次经过第一隐藏层、第一丢弃层、第二隐藏层和第二丢弃层,得到第四个分支的输出特征。
3.根据权利要求2所述的基于残差网络和序列特性的蛋白质分子识别特征预测方法,其特征在于,所述残差块a包含多个大小为3×3的卷积层,残差块a的输入特征与最后一个卷积层的输出特征进行跳跃连接,得到残差块a的输出特征;残差块b包含多个大小为3×3的卷积层,残差块b的输入特征经过1×1的卷积层后,再与最后个卷积层的输出特征进行跳跃连接,得到残差块b的输出特征。
4.根据权利要求1所述的基于残差网络和序列特性的蛋白质分子识别特征预测方法,其特征在于,第一个特征集包含16个序列特性,其中13个是从氨基酸指数的多种氨基酸特性中筛选得到的主要影响内源性无序蛋白质分子识别特征区域的氨基酸特性,剩余3个是反映蛋白质序列的结构特性,一个是待预测蛋白质序列的拓扑熵,另外两个是待预测蛋白质序列的氨基酸倾向性;第二个特征集和第三个特征集分别包含14和13个从氨基酸指数的多种氨基酸特性中筛选得到的主要影响内源性无序蛋白质分子识别特征区域的氨基酸特性;第四个特征集包含20个反映进化信息的序列特征,基于ncbi非冗余数据库的蛋白质序列并根据氨基酸的位置特异性得分矩阵筛选得到。
5.根据权利要求1或4所述的基于残差网络和序列特性的蛋白质分子识别特征预测方法,其特征在于,滑动窗口的长度分别为10、45和90。
技术总结本发明公开一种基于残差网络和序列特性的蛋白质分子识别特征预测方法,首先对蛋白质序列特性进行筛选,得到四个特征集,前三个特征集分别包括多个序列特性,第四个特征集包括多个反映进化信息的序列特性;然后,对待预测蛋白质序列进行预处理,提取待预测蛋白质序列各个残基关于每个特征集的特征矩阵;最后,构建预测模型;预测模型包含四个并行分支,四个分支的输入分别是残基关于四个特征集的特征矩阵,四个分支的输出经过全连接层,得到残基是否属于分子识别特征区域;对预测模型进行训练,将训练后的预测模型用于蛋白质分子识别特征区域的预测。该方法在待预测蛋白质序列预处理过程中采用双均值计算策略,细化了特征信息的局部变化和细微差异,实现了特征信息的充分提取,显著提高了预测精度。技术研发人员:何昊,贺瑾,周亚同,张智超,郝艺凡受保护的技术使用者:河北工业大学技术研发日:技术公布日:2024/10/17本文地址:https://www.jishuxx.com/zhuanli/20241021/318705.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。