基于氨基酸微环境与EMO神经网络的蛋白质设计方法
- 国知局
- 2024-07-11 17:33:36
本发明属于智能细胞生物识别领域,具体涉及一种基于氨基酸微环境与emo神经网络的蛋白质设计方法。
背景技术:
1、计算蛋白质设计(computational protein design,下称cpd)是指一种利用计算机算法和模型来预测和优化蛋白质结构和功能的方法,其目标是通过计算方法来寻找最优的蛋白质结构,以实现特定的生物学或应用目的。在过去的几十年里,cpd在酶工程、疫苗设计、抗体设计、膜蛋白设计、蛋白质交互作用等众多领域获得了巨大的成功。近期发表在nature的工作中,hongyuan lu等人利用mutcompute成功设计出了具有高稳定性和高水解活性的pet酶,该酶能在1周时间内水解51种pet塑料,有望解决困扰人类社会的塑料垃圾问题。持续的研究表明,cpd能在健康、医疗、环境、化工等各个领域造福人类社会。虽然cpd取得了显著的进展,但是如何精确地设计蛋白质的结构和功能仍然是一个巨大的挑战。
2、传统的cpd研究主要依据是anfinsen的折叠热力学假说。该假说的核心思想是:自然条件下,蛋白质总是折叠到自由能最低的构象。这也就意味着,蛋白质的3维结构完全是由其氨基酸序列所确定的。一个蛋白质序列对应了一种三维结构,但是相似的三维结构可能对应了多个蛋白质序列。传统cpd的主要研究内容就是从3维结构出发设计出自由能最低的序列。传统cpd方法的关注点有两个,其一是设计自由能计算函数,其二是寻找所需结构的最低能量氨基酸序列。基于能量函数的方法受限于本身能量函数的设计以及启发性算法寻优容易陷入局部最优的缺点,设计出的蛋白质往往达不到目标结构,使得蛋白设计实验成功率较低。
3、基于深度学习的cpd方法致力于从蛋白质氨基酸残基的周围化学结构特征中学习氨基酸残基与其周围结构的对应关系,这样就可以在不破坏蛋白质骨架的情况下,修改蛋白质的部分氨基酸,从而实现蛋白质设计。根据所使用的神经网络结构,这些方法主要分为基于多层感知机(multi-layer perceptron,下称mlp)的方法、基于卷积神经网络的方法以及基于图卷积神经网络的方法。但已经存在的方法仍然存在以下几点不足:(1)基于mlp的方法受限于手工特征的计算,通过经验设计出的手工特征有其局限性,这使得基于mlp方法的预测准确率陷入了瓶颈;(2)基于卷积神经网络的方法则是对氨基酸的原子三维环境特征进行建模。为了克服三维空间原子稀疏性,加速网络的收敛,该类方法均使用了高斯函数对三维环境特征进行了模糊处理。这种做法损失了部分信息,影响了最终的预测性能;(3)基于图神经网络的方法对原子环境建立图结构,优点是克服了原子在空间的稀疏性,缺点是忽略了原子的三维空间位置信息。因此,研究能从稀疏的原子环境中预测氨基酸种类从而设计蛋白质的方法依然是一个具有重要价值的挑战性工作。
技术实现思路
1、现有的cpd方法为了加快网络收敛采取了不同的处理方法,这样做损失了蛋白质的部分结构信息,在本发明中,搭建了一种新的神经网络模型,可以从稀疏的氨基酸微环境中识别出氨基酸种类,从而实现蛋白质的设计。该方法参考了emo模型中的逆残差移动模块,以及transformer模型中的多头自注意力机制,使得网络能关注稀疏的氨基酸微环境的重要组成部分,从而高效且精确的进行cpd任务。
2、本发明的技术方案如下:
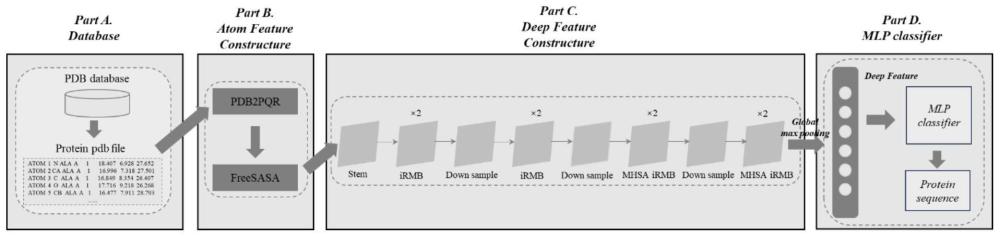
3、基于氨基酸微环境与emo神经网络的蛋白质设计方法,包括以下四个阶段:初始蛋白质原子特征构建,氨基酸微环境特征构建,深度特征构建,mlp分类器分类,4个阶段的步骤如下:
4、第一阶段:初始蛋白质原子特征构建。该阶段包含3个步骤,分别为给蛋白质pdb文件添加h原子,计算每个原子的自由电荷(free charge,下称fc)和溶剂可及表面积(solvent accessible surface area,下称sasa),以及构造每个原子的特征向量,其具体步骤如下:
5、第一步:使用pdb2pqr生物处理软件对目标蛋白质添加h原子,计算添加h原子后的每个蛋白质原子的fc;
6、第二步:使用freesasa生物处理软件计算每个蛋白质原子的溶剂可及表面积sasa;
7、第三步:将目标蛋白的每个原子别编码成7维特征向量,前5维表示o、c、n、s、h原子的one-hot编码,后2维表示原子的fc和sasa。;
8、第二阶段:氨基酸微环境特征构建。记目标蛋白的氨基酸数为m,从蛋白质的每个氨基酸原子特征向量获取氨基酸微环境的步骤如下:
9、第四步:对于非甘氨酸(gly),以氨基酸的cβ原子为中心,以氨基酸的n-cα键的方向为x轴方向,以垂直于n-cα-c平面且与cα-cβ键方向点积为正的方向为z轴方向,以与xz平面正交的方向为y轴方向,建立尺寸为的立方体3d网格,单元尺寸为
10、第五步:对于甘氨酸(gly),由于其没有cβ原子,其cβ的坐标由其余19种氨基酸n-cα键长以及n-cα-cβ键角计算而来,其余步骤与第四步相同;
11、第六步:去除中心氨基酸的r基团原子,每个单元网格被编码成落入其中原子的特征向量之和,一个氨基酸的微环境特征维度为7×20×20×20。
12、第七步:重复第一到三步,最终将目标氨基酸被编码成m个维度为7×20×20×20氨基酸微环境样本。
13、第三阶段:深度特征构建。构造深度特征需要经过3个步骤,分别为初步特征提取,浅层特征提取,深层注意力特征提取。
14、该阶段的具体步骤如下:
15、第一步:初步特征提取。将氨基酸微环境样本输入stem模块,获取初步特征f1;
16、stem模块中包括若干cna(convlayer,normlayer,active)基础模块,cna模块的结构如图2.a所示,给定输入ain,一个cna模块可以由以下公式表达:
17、fcna(ain)=factive(fnorm(fconv(ain))) (1)
18、式中fconv表示3d卷积层,fnorm是标准化处理,factive是指激活函数。
19、stem模块包含一个cna模块一个批标准化层、一个se模块、一个3d卷积层。给定输入ain,一个se模块可以由以下公式表达:
20、fse(ain)=fcna(fcna(fgmp(ain))) (2)
21、式中fgmp表示全局最大池化层。记b=fcna(fbn(ain)),那么stem模块可以表示为:
22、fstemm(ain)=ain+fconv(b+fse(b)·b) (3)
23、fbn表示批标准化层(batch_norm)。stem模块的第一个cna模块的标准化层为批标准化层,激活函数为silu;se模块中的两个cna模块分别使用relu和sigmoid激活函数。
24、第二步:浅层特征提取。将初步特征f1依次输入逆残差移动模块1、2,下采样模块1、逆残差移动模块3、4,下采样模块2共6个模块获得浅层特征f2;
25、逆残差移动模块(inverted residual mobile block,下称irmb)包含一个批标准化层、两个cna模块、一个3d卷积层。给定输入ain,记b=fcna(fbn(ain)),那么一个irmb模块可表示为:
26、firmb(ain)=ain+fconv(fcna(b)+b) (4)
27、下采样模块(downsample block)模块由一个批标准化层、两个cna模块、一个3d卷积层组成。给定输入ain,一个下采样模块可表示为:
28、fdown(ain)=fconv(fcna(fcna(ain))) (5)
29、irmb模块、down sample模块中包含的两个cna模块中,第一个使用relu激活函数且没有标准化层,第二个使用了使用silu激活函数和批标准化层。
30、第三步:深层注意力特征提取。将浅层特征f2依次输入多头自注意力逆残差移动模块1、2,下采样模块3,多头自注意力逆残差移动模块3、4,下采样模块4共6个模块获得深层注意力特征f3。
31、多头自注意力逆残差移动模块(multi-head self-attention irmb,下称mhsairmb)包含一个批标准化层、两个cna模块、一个多头自注意力机制模块,一个3d卷积层以及两个残差连接。给定输入ain,多头注意力机制可表示如下:
32、fmhsa(ain)=concat(h1,...,hh)wo (6)
33、式中,wo是对多头注意力得分的加权矩阵,hi是第i个自注意力得分,即:
34、
35、式中,记att=fmhsa(ain)·v,那么一个mhsa irmb模块可表示为:
36、fmhsa_irmb(ain)=fln(ain)+fconv(fcna(att)+att) (8)
37、mhsa irmb模块中包含的两个cna模块中,第一个使用relu激活函数且没有标准化层,第二个使用了使用silu激活函数和批标准化层。
38、第四阶段:mlp分类器分类。将深层注意力特征f3输入mlp分类器得到预测蛋白质氨基酸类别概率。
39、mlp分类器模块由一个全局最大池化层,一个平展层两个线性层以及之间的relu激活函数层组成。定输入ain,mlp分类器可表示如下:
40、fmlp_classifier(ain)=flinear(frelu(flinear(fflatten(fgmp(ain))))) (9)
41、式中,flinear是指线性层,frelu是relu激活函数层,fflatten是指平展层。
42、本发明的有益效果:
43、(1)区别于现有的方法使用模糊化处理蛋白质结构数据,本发明使用了多头自注意力机(multi-head self-attention,下称mhsa)制建立能关注稀疏氨基酸微环境的emo模型,解决了以往方法丢失信息和收敛慢的问题。
44、(2)本发明使用了逆残差移动模块(inverted residual mobile block,下称irmb),提高了氨基酸微环境的预测精度与预测效率。
45、(3)在两个广泛使用的数据集上,验证了本发明的优越性与有效性。
本文地址:https://www.jishuxx.com/zhuanli/20240615/84901.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表