一种含吡咯环的化合物的合成方法及其检测方法与流程
- 国知局
- 2024-09-05 14:25:59
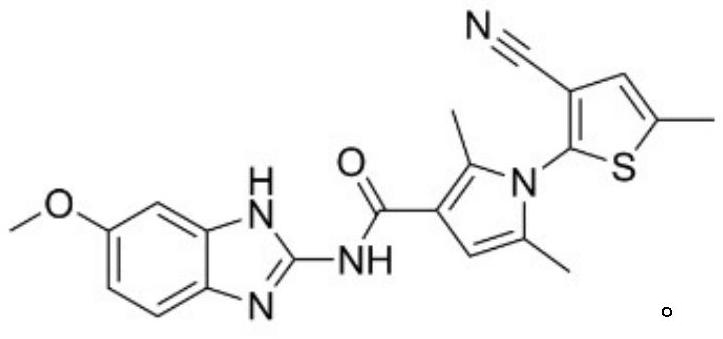
本发明涉及有机化合物的合成,具体涉及一种含吡咯环的化合物的合成方法,同时还涉及其检测方法。
背景技术:
1、dhx33属于含有dead/h盒的rna解旋酶蛋白家族,其中dead/h代表氨基酸asp-glu-ala-asp/his的缩写,这一序列连同其他多个保守性氨基酸序列出现在rna解旋酶家族成员的蛋白序列中,高度参与核酸底物结合以及atp水解。虽然这些家族成员共有这些相同的序列,但是每个rna解旋酶都有各自特定的专一性和独特的生物功能。人dhx33蛋白的分子量是72kda,具有解旋核酸的功能,它利用atp水解所释放的生物能来驱动改变rna和蛋白质复合物的构象,进而参与多种rna的代谢活动,具体而言,从rna转录、剪切、编辑、翻译到降解等一系列生物过程。dhx33的功能并不仅仅局限于对rna分子的修饰,研究表明,除了解旋rna双链之外,dhx33也参与dna的代谢。具体而言,dhx33可以解开dna的双链结构,并在基因表达过程中起重要作用。在体外的酶反应体系中,还发现dhx33可以解开dna/rna的杂合双链结构。
2、研究表明,dhx33通过结合多种癌症相关的基因启动子,影响了dna的甲基化状态,进而在基因组水平调控多种癌症基因的表达和肿瘤发展相关的信号通路,对细胞生长、增殖、迁移、凋亡、糖代谢等多种细胞活动有至关重要的作用。此外,发现dhx33可以感受外来双链rna分子的侵入并在细胞的先天免疫中发挥重要作用。dhx33作为十分重要的细胞生长调控基因,在多种癌症中高度表达。多种癌症的发生发展依赖于dhx33蛋白的高度表达。dhx33的遗传敲除可以显著抑制ras癌基因驱动的肺癌发生发展;体内和体外实验证实,抑制dhx33蛋白后,多种癌症如乳腺癌、结肠癌、脑胶质瘤、淋巴瘤等癌症的发生发展都受到明显抑制。研究还表明,dhx33的蛋白功能依赖于其解旋酶活力。dhx33的解旋酶活力缺失突变体不具有dhx33蛋白的功能,无法替代野生型dhx33基因的功能。
3、目前已有专利公开了dhx33的抑制剂,例如多环化合物ky386,其结构式为:
4、
5、ky386主要的合成路线如下所示:
6、
7、其中,化合物5是合成ky386的中间体,该化合物与化合物7缩合之后即可获得ky386化合物。化合物5的合成分两步进行:第一步先制备化合物3,即2-乙酰基-4-戊酮酸乙酯,是以化合物1(乙酰乙酸乙酯)为起始原料,与化合物2(氯丙酮)反应制得,该步骤产率低于20%;第二步通过化合物3与化合物4(2-氨基-3-氰基-5-甲基噻吩)反应,制得化合物5,该步骤产率约40%。
8、以上多环化合物ky386的中间体化合物5的制备工艺收率低,且制得的化合物5为黄色油状物,产物提纯分离困难,需要使用柱色谱进行纯化分离,耗时长,成本大;最重要的是,油状化合物不易开展杂质控制和严格的质量分析,不利于后期临床药物的有效质量控制。
9、基于以上问题,本发明旨在提供一种含吡咯环的固体化合物的合成方法。
技术实现思路
1、有鉴于此,本发明的目的在于提供一种含吡咯环的化合物的合成方法,用以解决现有ky386合成中中间体制备收率低且纯化难的问题。
2、此外,本发明的目的还在于提供一种制得的含吡咯环的化合物的检测方法。
3、为实现上述目的,本发明采用的技术方案为如下所述。
4、第一方面,本发明提供了一种含吡咯环的化合物的合成方法,所述含吡咯环的化合物的结构式如i-4所示:
5、
6、合成方法包括以下步骤:使式i-2所示的化合物与式i-3所示的化合物反应,以生成式i-4所示的化合物;
7、
8、在上述技术方案的一些实施方式中,所述反应在催化剂作用下进行,所述催化剂选自醋酸、硫酸、对甲苯磺酸、三氯化铝。所述催化剂可以选自醋酸、硫酸、对甲苯磺酸、三氯化铝中的任一种,或者是两种或以上的组合。
9、在上述技术方案的一些实施方式中,所述反应在有机溶剂中进行,所述有机溶剂选自四氢呋喃、含苯环溶液、乙腈、二氯甲烷。
10、其中,所述含苯环溶液选自苯、c7~9的液态取代苯;优选地,所述含苯环溶液选自甲苯、二甲苯、三甲苯、乙苯、丙苯、异丙苯。
11、在上述技术方案的一些实施方式中,式i-2所示的化合物与式i-3所示的化合物反应的反应温度为30℃~130℃。
12、在上述技术方案的一些实施方式中,所述式i-2所示化合物与所述式i-3所示化合物的投料摩尔比为0.8~1.2,优选为0.8~0.9,进一步优选为0.8~0.85。
13、在上述技术方案的一些实施方式中,所述催化剂与所述式i-3所示化合物的投料摩尔比为0.1~0.5,优选为0.1~0.3,进一步优选为0.18~0.20。
14、在上述技术方案的一些实施方式中,所述式i-2所示化合物的合成方法包括以下步骤:使式i-1所示化合物与氯丙酮反应,以生成式i-2所示的化合物;
15、
16、在上述技术方案的一些实施方式中,式i-1所示化合物与氯丙酮反应的反应温度为5℃~30℃。
17、在上述技术方案的一些实施方式中,式i-1所示化合物与氯丙酮反应的反应时间为8~16小时。
18、在上述技术方案的一些实施方式中,所述反应在添加氢化钠的条件下进行。
19、在上述技术方案的一些实施方式中,所述反应在有机溶剂中进行,所述有机溶剂选自四氢呋喃、含苯环溶液、乙腈、二氯甲烷;其中,所述含苯环溶液选自苯、c7~9的液态取代苯,优选地,所述含苯环溶液选自甲苯、二甲苯、三甲苯、乙苯、丙苯、异丙苯。
20、在上述技术方案的一些实施方式中,所述式i-1所示化合物与所述氯丙酮的投料摩尔比为0.8~1.3,优选为1.0~1.3,进一步优选为1.2~1.3。
21、在上述技术方案的一些实施方式中,所述式i-1所示化合物与所述氢化钠的质量比为7~10,优选为8~10,进一步优选为8~9。
22、第二方面,本发明提供了一种式i-4所示化合物的检测方法,采用高效液相色谱检测方法进行检测,检测条件包括:采用c18反相色谱柱,检测波长为274nm;
23、以流动相a和流动相b进行梯度洗脱,所述流动相a为体积百分比浓度0.1%的甲酸溶液,所述流动相b为甲醇;梯度洗脱程序为:
24、
25、之后,依据式i-4所示化合物的峰面积使用面积归一法计算出纯度。
26、本发明提供的式i-4所示化合物的检测方法可以实现检测目标化合物式i-4所示化合物与其他杂质和未反应的起始原料的有效分离。
27、本发明合成方法具有如下优点:
28、本发明采用了新的合成路线,获得的中间体即所述式i-4所示化合物为固体,易于分离纯化和质量控制。本发明方法合成的收率高,且合成获得的式i-4所示化合物粗产物纯度高,无需色谱柱纯化。因为不需要开展繁琐的柱色谱分离纯化,因此有效降低了生产成本。
29、采用本发明提供的式i-4所示化合物,与化合物7发生脱水缩合反应,可以一步制得化合物ky386。以本发明合成的式i-4所示化合物代替现有制备工艺中的化合物5与化合物7发生反应制备ky386,可以有效规避油状化合物作为中间体的不利后果,利于有效开展ky386临床药物的质量控制。
30、
31、上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,而可依照说明书的内容予以实施,并且为了让本发明的上述和其他目的、特征和优点能够更明显易懂,以下特举较佳实施例,并配合附图,详细说明如下。
本文地址:https://www.jishuxx.com/zhuanli/20240905/286465.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。