一种基于杂交捕获测序的缺失型α-地中海贫血基因类型的检测方法与流程
- 国知局
- 2024-10-09 14:37:55
本发明属于分子生物学,具体涉及一种基于杂交捕获测序技术检测缺失型α-地中海贫血基因类型的方法。
背景技术:
1、地中海贫血(thalassaemia,简称地贫)是指由珠蛋白基因缺陷(突变、缺失)导致的一种或多种珠蛋白肽链合成障碍引起的遗传性溶血性贫血,是临床上最常见的单基因遗传病之一。
2、α-地中海贫血(α-地贫)是由于α珠蛋白肽链的合成受到部分或完全抑制,导致血红蛋白(hb)合成不足而引发的遗传性溶血性贫血,呈常染色体隐性遗传,致病基因为hba1和hba2,男女患病几率相等。α-地贫是世界范围内高发的单基因遗传病,大约5%的世界人口携带α-珠蛋白基因的变异。α-地贫突变又分为缺失型和非缺失型,其中缺失型突变占大多数。根据缺失的α基因数目,可分为α+地贫(缺失1个α基因,-α/)和α0-地贫(缺失2个α基因,--/)。其中,有6种α-基因突变(--sea/、-α3.7/、-α4.2/、hba2:c.369c>g、hba2:c.427t>c、hba2:c.377t>c)占据了中国人群总数的98%。
3、目前,对于α-地贫,临床采用的诊断策略均是先用一线技术检测高发的已知突变,若无法确诊,再采用二线技术检测罕见或未知突变。对于点突变,一线技术为rdb法或pmca法,二线技术为sanger测序;对于缺失突变,一线技术为gap-pcr法,二线技术为mlpa法。这些检测方法中,rdb法和pmca法均只能用于检测已知突变,而sanger测序通量低、测序成本较高;gap-pcr借助多个引物的组合,可以根据扩增产物区分野生型、突变杂合子和突变纯合子,是目前检测缺失的常规方法,但其局限性是需要根据缺失的范围设计引物,仅能检测已知突变;mlpa可同时检测已知和未知的缺失突变,但需配备专用的仪器,且试剂盒成本较高。
4、因此,开发一种缺失型α-地贫基因类型检测方法为市场所需,本发明利用杂交捕获测序技术,快速而高效地检测地中海贫血相关基因的变异情况;通过标准化处理和差异分析,获得每个捕获区间的log2 ratio,使得数据更加准确可靠。采用局部加权回归算法对数据进行处理,有效地降低了数据的噪音,并且平滑出基因型变异的趋势,提高了数据的可靠性和解读的一致性。该方法可以对地中海贫血相关基因的整体变异情况进行全面分析,为突变分类和变异检测提供更可靠的数据基础,为地中海贫血等遗传性疾病的筛查提供重要的遗传学信息支持。
技术实现思路
1、为解决上述问题,本发明通过为杂交捕获测序技术配备一套合适的算法,提供一种准确率高、成本效益高的缺失型α-地贫地中海贫血基因类型检测方法。
2、为达到上述目的,本发明的技术方案如下:
3、本发明提供一种基于杂交捕获测序的缺失型α-地中海贫血基因类型的检测方法,包括以下步骤:
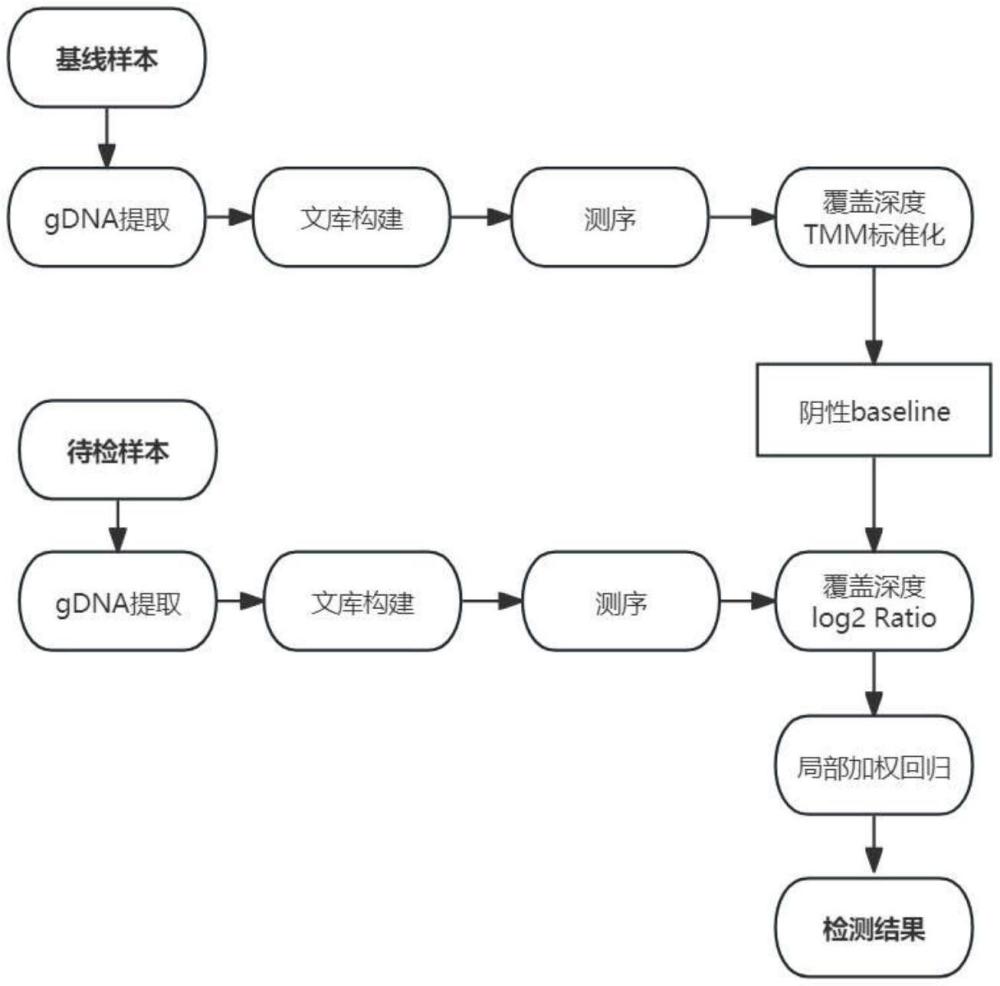
4、步骤一:gdna提取,收集受试者静脉血、口腔拭子、唾液任意方式的样本(包括基线样本和待检样本),根据不同的组织类型选择相应的核酸提取试剂盒进行gdna的提取,质控标准为保证dna提取总量≥500ng,a260/a280=1.8-2.0;
5、步骤二:文库构建和测序,对质控合格的gdna样本进行测序文库构建,保证该测序文库的插入片段集中在250-300bp,确保文库质控合格;使用探针panel对合格文库进行目标区域的杂交和捕获富集;然后对富集后的文库进行pe150测序;获得原始下机数据,qc指标合格,平均测序深度≥150x;
6、步骤三:测序数据计算和tmm标准化,使用bedtools软件计算基线样本每个捕获区间的覆盖深度;使用20个基线样本每个捕获区间的覆盖深度建立baseline,并进行tmm标准化;使用bedtools软件计算待检样本每个捕获区间的覆盖深度,定义为coverage;对baseline和coverage进行合并,再次进行tmm标准化。
7、步骤四:数据分析,使用r语言deseq2工具包对步骤三合并后的数据进行差异表达分析,得到基线样本和待检样本每个捕获区间的log2 ratio(差异倍数)信息;
8、步骤五:平滑曲线图的绘制,对基线样本和待检样本每个捕获区间的log2 ratio数值执行局部加权回归算法,平滑得出基因型变异的趋势,进而判断地贫突变的基因类型。
9、进一步地,步骤一中所述的受试者样本包括静脉血、口腔拭子、唾液任一方式的样本。
10、进一步地,所述缺失型α-地中海贫血基因类型具体包括:-α3.7/,-α4.2/,--sea/缺失类型。
11、进一步地,步骤三中,tmm标准化具体包括以下步骤:
12、s1-1:首先,计算每个样本的比例因子,比例因子用于调整每个样本的覆盖深度,以使每个样本的覆盖深度在总体上保持一致;
13、s1-2:其次,去除比例因子中的异常值,当比例因子低于中位数减去1.5倍iqr或高于中位数加上1.5倍iqr的值被视为异常值;
14、s1-3:然后,将每个捕获区间的覆盖深度与相应的比例因子相乘来进行标准化;
15、s1-4:最后,检查执行标准化后的效果,以确保每个样本的覆盖深度在总体上达到平衡。
16、进一步地,待检样本平滑曲线图的绘制具体步骤如下:
17、步骤a:利用bedtools软件分别计算待检样本每个捕获区间的覆盖深度;
18、步骤b:将待检样本覆盖深度数据和baseline数据合并,然后分别进行tmm标准化处理;
19、步骤c:利用r语言中的deseq2工具包对步骤b合并后的数据分别进行差异表达分析,从而获得待检样本每个捕获区间的log2 ratio(差异倍数)信息;
20、步骤d:最后,对待检样本每个捕获区间的log2 ratio数值执行局部加权回归算法,分别平滑出基因型变异的趋势。
21、步骤e:根据获得的平滑曲线图判断待检样本的地贫基因类型。
22、在本发明的实施方案中,缺失型α-地贫基因类型的判读标准为:根据绘制出的平滑曲线(横轴为x轴,纵轴为y轴)所在的区间和差异倍数,判断目标基因突变的类型。判断原则为:如平滑曲线位于y=0上或其附近,则定义为阴性样本;如平滑曲线存在一个极小值(y<0),且该值位于虚线b1/b2区域内,则定义该基因突变为-α4.2/缺失型;如平滑曲线存在一个极小值(y<0),且该值位于虚线c1/c2区域内,则定义该基因突变为-α3.7/缺失型;如平滑曲线在虚线a1/a2区域内的y值恒小于0,则定义该基因突变为--sea/缺失型。
23、本发明的有益效果为:
24、(1)本发明基于杂交捕获测序技术检测缺失型α-地中海贫血相关基因的变异情况,快速且高效。
25、(2)本发明利用局部加权回归算法处理数据,有效地降低了数据的噪音,平滑出基因型变异的趋势,有助于提高数据的可靠性和解读的一致性。
26、(3)本发明适用于血液等多种类型的样本检测,具有广泛的应用前景,有助于为地中海贫血等遗传性疾病的筛查提供重要的遗传学信息支持。
本文地址:https://www.jishuxx.com/zhuanli/20241009/305912.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表