7H-吡咯并[2,3-d]嘧啶-4-胺衍生物的制造方法与流程
- 国知局
- 2024-12-06 12:26:20
本发明涉及7h-吡咯并[2,3-d]嘧啶-4-胺衍生物的制造方法以及能够大量制造的7h-吡咯并[2,3-d]嘧啶-4-胺衍生物等。
背景技术:
1、在工业制造医药品有效成分(active pharmaceutical ingredients;以下也称为api)时,从原料制造api的工序要求工序数少且收率高。另外,api的制造中要求能够大量制造,因此,不希望有不适合于大量制造的操作。并且,通过这样的工序制造的api的品质必须符合人用药品技术要求国际协调理事会(以下也称为ich)所规定的基准等,作为基准的对象可以列举api所含杂质、残留溶剂和残留金属等。
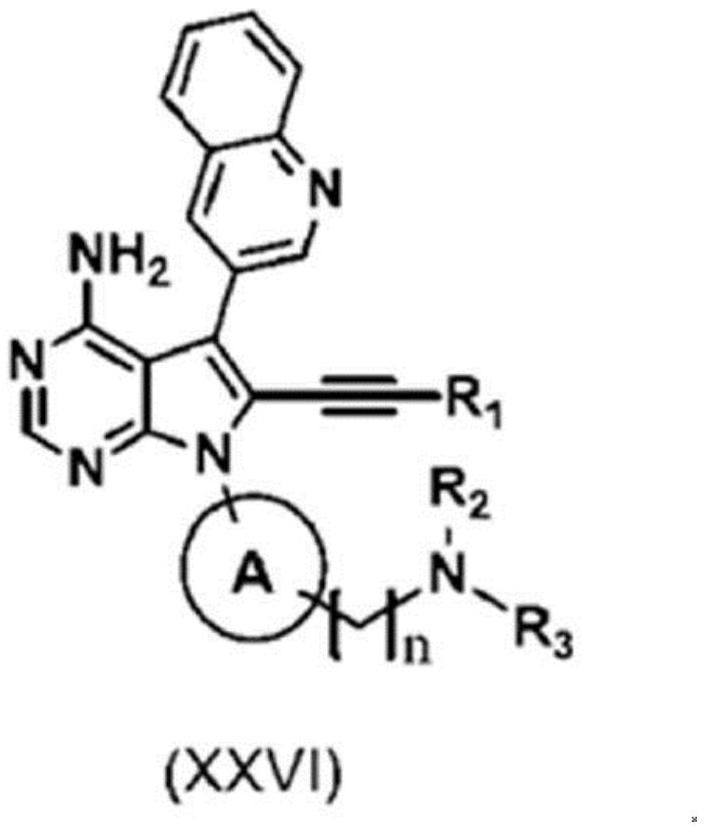
2、专利文献1中,作为具有egfr抑制作用的化合物,公开了n-(4-(4-氨基-6-乙炔基-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-甲酰胺(以下,也称为化合物(1))。并且,作为其制造方法的一例,记载了如下路径:通过使式(xxii)所示的化合物和式(xxiii)所示的化合物通过薗头偶联(sonogashira coupling)进行反应,由此导入r1-c≡c-基(r1为氢原子或任选地具有取代基的c1-c3烷基)而得到式(xxiv)所示的化合物,接着,通过胺的脱保护,转换为式(xxv),最后通过实施酰基化反应,合成式(xxvi)的化合物。
3、
4、现有技术文献
5、专利文献
6、专利文献1:国际公开wo2020/166680号小册
技术实现思路
1、发明所要解决的课题
2、但是,作为化合物的合成路径,在进一步研究适于大量生产的方法的方面尚有余地。在这样的状况下,寻求一种适于作为api大量合成的化合物(1)或者后述的式(i)所示的化合物的制造方法。特别是寻求适于作为api大量合成的特性之中,杂质的混入少和收率高之中的一个以上的特性良好的化合物(1)或者后述的式(i)所示的化合物的制造方法。
3、用于解决课题的方法
4、本发明的发明人进行了深入研究,结果完成了通过在碱性条件下将末端乙炔的保护基脱保护,从而适于大量合成的具有一个以上的特性的式(i)所示的化合物的制造方法。
5、即,本发明提供例如如下的[1]~[14]。
6、[1]一种式(i)所示的化合物或其盐的制造方法,其包括:
7、通过在碱性条件下,将式(ii)的r1所示的保护基脱保护,形成式(i)所示的化合物或其盐的步骤,
8、
9、(在上述式(i)和式(ii)中,
10、r1表示保护基,
11、r2表示任选地具有保护基的氨基(胺)、卤素原子或离去基团,
12、r3表示任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基)。
13、[2]如[1]所述的制造方法,其中,在上述式(i)和式(ii)中,r1表示硅连接保护基,r2表示氨基(胺),r3表示任选地具有甲基的吡嗪基。
14、[3]如[1]或[2]所述的制造方法,其中,上述式(i)所示的化合物为n-(4-(4-氨基-6-乙炔基-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-甲酰胺或其盐。
15、[4]一种式(i)所示的化合物或其盐的制造方法,其包括:
16、(1)通过式(iii-1)所示的化合物与式(viii)所示的化合物的连接,衍生为式(iii-2)所示的化合物的步骤;或通过式(iii-1)所示的化合物与式(iv)所示的化合物的连接,制造式(v-1)所示的化合物的步骤;
17、
18、
19、(2)通过式(iii-2)所示的化合物与式(iv)所示的化合物的连接,或通过式(v-1)所示的化合物与式(viii)所示的化合物的连接,衍生为式(ii)所示的化合物的步骤;和
20、
21、(3)通过在碱性条件下将式(ii)的r1所示的保护基脱保护,形成式(i)所示的化合物或其盐的步骤,
22、
23、(在上述式(i)、(ii)、(iii-1)、(iii-2)、(iv)、(v-1)和(viii)中,
24、r1表示保护基,
25、r2表示任选地具有保护基的氨基(胺)、卤素原子或离去基团,
26、r3表示任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基,
27、r5和r5’分别独立地表示氢原子或氨基(胺)的保护基,或者nr5r5’表示邻苯二甲酰亚胺或硝基,
28、x表示卤素原子或离去基团)。
29、[5]如[4]所述的制造方法,其中,
30、在上述式(i)、(ii)、(iii-1)、(iii-2)、(iv)和(v-1)中,
31、r1表示硅连接保护基,
32、r2表示氨基(胺),
33、r3表示任选地具有甲基的吡嗪基,
34、r5表示氢原子或boc,r5’表示氢原子。
35、[6]如[4]或[5]所述的制造方法,其中,
36、上述式(i)所示的化合物为n-(4-(4-氨基-6-乙炔基-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-甲酰胺或其盐。
37、[7]如[4]所述的制造方法,其中,
38、使式(vi)所示的化合物与式(vii)所示的化合物反应,得到式(ix)的化合物的步骤,和
39、由式(ix)的化合物得到式(iii-1)所示的化合物的步骤,
40、
41、(式中,l表示卤素原子或离去基团)
42、
43、(在式(vi)和式(ix)中,
44、r4表示nr5r5’,
45、r5和r5’分别独立地表示氢原子或氨基的保护基,或者nr5r5’表示邻苯二甲酰亚胺或硝基,
46、l表示卤素原子或离去基团)。
47、[8]如[7]所述的制造方法,其中,
48、在所述式(vi)、(vii)和(ix)中,
49、r4表示nr5r5’,
50、r5表示boc,r5’表示氢原子,
51、l表示氯原子。
52、[9]如[4]~[8]中任一项所述的制造方法,其中,
53、不包括利用柱色谱法的精制。
54、[10]如[1]~[9]中任一项所述的制造方法,其中,
55、在通过上述方法制得的上述式(i)所示的化合物及其盐中,式(i-1)所示的化合物的含量为0.2%以下,
56、
57、(式中,
58、r3表示任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基)。
59、[11]一种组合物,其是由[1]~[10]中任一项所述的方法得到的、包含式(i)所示的化合物或其盐的组合物。
60、[12]一种式(ii)所示的化合物或其盐,
61、
62、(式中,
63、r1表示保护基,
64、r2表示任选地具有保护基的氨基、卤素原子或离去基团,
65、r3表示任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基)。
66、[13]如[12]所述的化合物或其盐,其中,
67、在上述式(ii)中,r1表示硅连接保护基,r2表示氨基,r3表示任选地具有甲基的吡嗪基。
68、[14]如[12]或[13]所述的化合物或其盐,其中,
69、上述式(ii)所示的化合物为n-(4-(4-氨基-5-(喹啉-3-基)-6-((三乙基甲硅烷基)乙炔基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-羧酰胺或其盐。
70、发明的效果
71、根据本发明的一个方式的制造方法,能够提供适于作为api大量合成的特性之中,杂质的混入少和收率高之中的一个以上的特性良好的化合物(1)或者后述的式(i)所示的化合物的制造方法。根据本发明的优选方式的制造方法,能够提供还具有不使用含氟试剂、不使用爆炸性试剂和能够避免利用柱色谱法的精制之中一个以上的特性的制造方法。
72、根据本发明的一个方式,能够提供通过在碱性条件下将式(ii)所示的化合物的r1所示的保护基脱保护而无需进行柱色谱法的精制来制造式(i)所示的化合物或其盐的方法。而专利文献1所述的方法需要利用硅胶柱色谱法进行精制。
73、
74、(其中,r1表示保护基,r2表示任选地具有保护基的氨基、卤素原子或离去基团,r3表示任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基)
75、
76、(其中,r2和r3与上述相同。)
77、在下文中记载由式(ii)向式(i)衍生的工序。
78、在本说明书中,r1表示保护基,是用于保护式(i)中的末端乙炔的氢原子的取代基。末端乙炔是指乙炔中的2个氢原子之中的一个被取代的乙炔,是指式(i)中的-c≡ch(也称为乙炔基)。在本发明的一个实施方式中,r1只要是能够在碱性条件下脱保护的保护基即可,能够使用任意的保护基。作为这样的r1中的保护基,可以列举将末端乙炔的氢原子取代为碳原子的保护基(碳连结保护基)、取代为硅原子的保护基(硅连接保护基)、取代为磷原子的保护基(磷连结保护基)等。
79、在本说明书中,作为碳连结保护基,可以列举叔丁基、环烷基、2-羟基丙烷-2-基和二甲基氨基甲基等,优选为2-羟基丙烷-2-基。
80、在本说明书中,作为硅连接保护基,例如可以列举tms(三甲基甲硅烷基)、tes(三乙基甲硅烷基)、tbs(叔丁基二甲基甲硅烷基)、tips(三异丙基甲硅烷基)和tbdps(叔丁基二苯基甲硅烷基)等。优选为tes或tips,更优选为tes。
81、在本说明书中,作为磷连结保护基,可以列举-pph2(二苯基膦)和-p(o)ph2等,优选为-p(o)ph2(ph表示苯基)。
82、本发明的一个实施方式中的r1由于是能够在碱性条件下进行脱保护的保护基,能够在脱保护的同时将可能在反应工序中作为副产物生成的式(i-1)所示的化合物等的类似物转换为目的化合物。通过该转换,能够降低杂质的混入,并且为高收率,因此,能够实现作为医药品原料的高纯度化。
83、
84、(其中,r3与上述相同。)
85、在本发明的一个实施方式中,r1优选为选自碳连结保护基、硅连接保护基和磷连结保护基中的保护基,更优选为硅连接保护基,进一步优选为tms(三甲基甲硅烷基)、tes(三乙基甲硅烷基)、tbs(叔丁基二甲基甲硅烷基)、tips(三异丙基甲硅烷基)和tbdps(叔丁基二苯基甲硅烷基),特别优选为tes(三乙基甲硅烷基)。
86、本发明的一个实施方式所涉及的制造方法中使用的化合物的r2为任选地具有保护基的氨基、卤素原子或离去基团。
87、作为r2中的“任选地具有保护基的氨基”的保护基,没有特别限制,例如能够列举叔丁氧基羰基(boc)、苄氧基羰基(cbz或z)、9-芴基甲氧基羰基(fmoc)、烯丙氧基羰基(alloc)、2,2,2-三乙氧基羰基(troc)等氨基甲酸酯保护基、邻硝基苯磺酰基(o-ns、ns、nitrobenzenesulfonyl或o-nitrobenzenesulfonyl)、对硝基苯磺酰基(p-ns或p-nitrobenzenesulfonyl)、2,4-二硝基苯磺酰基(dns)、ses(2-三甲基甲硅烷基乙烷磺酰基)等磺胺保护基、r2为邻苯二甲酰(phth)的保护基,优选为氨基、具有氨基甲酸酯保护基的氨基、和邻苯二甲酰亚胺,更优选为氨基、以及具有1或2个选自boc、cbz、fmoc、alloc和troc中的一个保护基的氨基,更优选为氨基、以及具有1或2个选自boc、cbz、和fmoc中的一个保护基的氨基,特别优选为氨基。
88、作为r2中的卤素原子,可以列举氟原子、氯原子、溴原子和碘原子,优选为氯原子。
89、作为r2中的离去基团,可以列举-oso2cnfn+2(n表示1~4的整数)、-so2cnfn+2、甲磺酸基(-oms;ms表示甲磺酰基)、甲磺酰基、甲苯磺酸基(-ots;ts表示对甲苯磺酰基)、对甲苯磺酰基、硝基苯磺酸基(-ons或-op-ns)、对硝基苯磺酰基、-oso2ph(ph表示苯基)、-so2ph、苯氧基(-oph)等,优选为甲磺酰基。或者,离去基团可以为-oso2cnf2n+1(n表示1~4的整数)、-so2cnf2n+1、甲磺酸基(-oms;ms表示甲磺酰基)、甲磺酰基、甲苯磺酸基(-ots;ts表示对甲苯磺酰基)、对甲苯磺酰基、硝基苯磺酸基(-ons或-op-ns)、对硝基苯磺酰基、-oso2ph(ph表示苯基)、-so2ph、苯氧基(-oph)等。
90、本发明的一个实施方式所涉及的制造方法中使用的化合物的r2为任选地具有保护基的氨基、卤素原子或离去基团,或者r2为邻苯二甲酰亚胺。优选为氨基、具有1或2个选自boc、cbz、fmoc、alloc、troc、和ns中的一个保护基的氨基、氟原子、氯原子、溴原子、碘原子、-oso2cnfn+2(n表示1~4的整数)、-so2cnfn+2、甲磺酸基、甲磺酰基、甲苯磺酸基、p-甲苯磺酰基、硝基苯磺酸基、p-硝基苯磺酰基、-oso2ph、-so2ph、和苯氧基,或r2为邻苯二甲酰亚胺,更优选为氨基、boc、cbz、fmoc、氯原子、和甲磺酰基,更优选为氨基、氯原子、和甲磺酰基,特别优选为氨基。或者,r2为氨基、具有1或2个选自boc、cbz、fmoc、alloc、troc和ns中的一个保护基的氨基、氟原子、氯原子、溴原子、碘原子、-oso2cnf2n+1(n表示1~4的整数)、-so2cnf2n+1、甲磺酸基、甲磺酰基、甲苯磺酸基、对甲苯磺酰基、硝基苯磺酸基、对硝基苯磺酰基、-oso2ph、-so2ph、和苯氧基,或r2为邻苯二甲酰亚胺。
91、在本发明的一个实施方式中,式(i)或式(ii)所示的化合物的r3为任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基。作为“任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基”,优选为具有选自氮原子、氧原子和硫原子的1~4个杂原子的5~10元的单环式或者多环式的完全不饱和或部分饱和的杂环基,更优选为哒嗪基、嘧啶基、噁唑基、噁二唑基、吡嗪基、吡啶基、咪唑基、呋喃基、异噁唑基、三唑并吡啶基、三唑基、三嗪基、噻唑基、噻二唑基、咪唑并吡嗪基、或吡唑基,更进一步优选为异噁唑基、吡唑基、噁唑基、噁二唑基、吡嗪基、或哒嗪基,更加进一步优选为吡唑基、噁唑基、噁二唑基、吡嗪基、或哒嗪基,特别优选为吡嗪基。
92、在本发明的一个实施方式中,作为r3所示的“任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基”的“取代基”,为卤素原子、氰基、氧基、c1~c6烷基、c1~c6卤代烷基、c1~c6烷氧基-c1~c6烷基、c1~c6烷氧基、c1~c6卤代烷氧基、c1~c6单或者二烷基氨基、c1~c6烷基磺酰基、c3~c7环烷基、或c6~c10芳香族烃。优选为氟原子、氯原子、氰基、氧基、c1~c6烷基、单氟代甲基、甲氧基乙基、甲氧基、单氟代甲氧基、二甲基氨基、甲基磺酰基、环烷基、或苯基,更优选为c1~c6烷基,更进一步优选为甲基。在本说明书中,取代基的数量可以为1个也可以为多个。
93、在本发明的一个实施方式中,r3为任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基,优选为任选地具有取代基的具有选自氮原子、氧原子和硫原子中的1~4个杂原子的5~10元的单环式或者多环式的完全不饱和或部分饱和的杂环基,更优选为任选地具有取代基的哒嗪基、嘧啶基、噁唑基、噁二唑基、吡嗪基、吡啶基、咪唑基、呋喃基、异噁唑基、三唑并吡啶基、三唑基、三嗪基、噻唑基、噻二唑基、咪唑并吡嗪基或吡唑基,更优选为任选地具有取代基的吡嗪基,更优选为任选地具有c1~c6烷基的吡嗪基,更优选为具有甲基的吡嗪基,更优选为5-甲基吡嗪-2-基。
94、在本发明的优选实施方式中,式(i)所示的化合物为n-(4-(4-氨基-6-乙炔基-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-甲酰胺。如专利文献1也所示的那样,该化合物为具有优异的抑制活性的化合物,故而优选。
95、在本发明的一个实施方式中,由式(ii)所示的化合物衍生为式(i)所示的化合物时产生的副产物只要是式(i)所示的化合物以外的化合物即可,没有特别限制,优选为在碱性条件下转换为式(i)所示的化合物的副产物,更优选为式(i-1)所示的化合物。
96、
97、(式中,r3的定义和实施方式与上述相同)
98、例如,r3为5-甲基吡嗪-2-基时,式(i-1)所示的化合物为n-(4-(6-乙炔基-4-(5-甲基吡嗪-2-羧酰胺)-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-羧酰胺(以下也称为化合物(2)),具有以下结构。
99、
100、在本发明中,碱性条件下是指通过碱将反应体系中调整为碱性的状态。作为该碱,只要反应进行即可,没有特别限制,例如,作为无机碱,能够列举金属氢氧化物(氢氧化钠、氢氧化钙等)、金属氢化物(氢化锂、氢化钠等)、金属碳酸盐(碳酸钠、碳酸钾、碳酸铯、碳酸钙、碳酸锂、碳酸镁、碳酸氢钠等)等,作为有机碱,能够列举金属醇盐(甲醇钠、叔丁醇钾等)、金属酰胺(氨基钠、二异丙氨基锂等)、烷基金属化合物(正丁基锂、三甲基铝等)、烷基胺(三乙胺、四甲基乙二胺、哌啶、1,4-二氮杂双环[2.2.2]辛烷等)、杂环胺类(二氮杂双环十一烷、吡啶、咪唑等)和氟化季铵(四正丁基氟化铵)等。优选为不含氟离子的碱,更优选为不含氟化物离子的金属碳酸盐或含钾的盐,进一步优选为碳酸钾。能够将这些单独或组合而用于本发明的制造方法。
101、关于该试剂的使用量,只要反应进行即可,没有特别限制,例如相对于式(ii)所示的化合物1摩尔,能够使用0.1~50摩尔。优选为0.1~10摩尔、进一步优选为0.1~2摩尔。
102、关于脱保护工序的溶剂,只要能够利用碱将反应体系调整为碱性且能够将反应所使用的化合物溶解即可,没有特别限制。例如,可以列举c5~c10烃、c6~c14芳香族烃、c1~c6醇、c3~c10脂肪族碳酸酯、c3~c10酮、c4~c10醚、非质子性剂型有机溶剂,或这些的混合溶剂。此外,在本说明书中,以c数字表示的记载是指碳原子数,是指这样的化合物中所含的碳原子的总数。
103、c5~c10烃是碳原子数为5~10的烃,可以列举戊烷、己烷、庚烷、辛烷、壬烷、癸烷、环戊烷、环己烷、环庚烷、环辛烷、环壬烷、环癸烷等。
104、c6~c14芳香族烃是碳原子数为6~14的芳香族烃,可以列举苯、萘、蒽、甲苯、二甲苯、枯烯、苯乙烯、菲等。
105、c1~c6醇是碳原子数为1~6的醇,可以列举甲醇、乙醇、正丙醇、异丙醇、正丁醇、叔丁醇、正戊醇、正己醇等。
106、c3~c10脂肪族碳酸酯是碳原子数为3~10的脂肪族碳酸酯,可以列举甲酸丙酯、乙酸甲酯、乙酸乙酯、乙酸丙酯、丙酸甲酯、丙酸乙酯、丙酸丙酯、丁酸甲酯、丁酸乙酯等。
107、c3~c10酮是碳原子数为3~10的酮,可以列举丙酮、乙甲酮、二乙酮、异丙基甲基酮、环己酮等。
108、c4~c10醚是碳原子数为4~10的醚,可以列举二乙醚、叔丁基甲基醚、四氢呋喃、1,4-二噁烷等。
109、非质子性极性有机溶剂是指不存在离子化的质子的溶剂,可以列举n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、氯仿、二氯甲烷等。
110、上述溶剂之中,优选为二氯甲烷、甲醇、乙醇、n-甲基吡咯烷酮、进一步优选为二氯甲烷、甲醇。另外,溶剂量只要是使反应进行即可,没有特别限制,相对于式(ii)的化合物1kg,优选为10~100l,进一步优选为15~50l,更进一步优选为20~30l。
111、脱保护工序的反应时间只要是能够实现脱保护的时间即可,没有特别限制,能够适当设定。例如为1~120小时、优选为6~48小时、进一步优选为12~24小时。
112、脱保护工序的反应温度只要是能够实现脱保护的温度即可,没有特别限制,能够适当设定。例如为0~60℃、优选为10~50℃、进一步优选为20~40℃。(需要说明的是,在本发明中,室温是指18~25℃。)
113、另外,根据本发明的其它方式,可以提供式(ii)所示的化合物。式(ii)所示的化合物除了制造式(i)所示的化合物以外,也是有用的中间体。在本发明的一个实施方式中,作为式(ii)所示的化合物中的r1、r2和r3,能够适当组合上述的任意选择。
114、<反应通路>
115、本发明的一个实施方式的制造方法包括:
116、(1)使式(iv)所示的化合物与式(iii)所示的化合物反应,制造式(v)所示的化合物的工序,
117、
118、(在上述式(iii)~式(v)中,
119、r1表示保护基,
120、r2表示任选地具有保护基的氨基、卤素原子或离去基团,
121、r3表示任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基,
122、r4表示nr5r5’或nhcor3,r5和r5’分别独立地表示氢原子、或氨基的保护基,或者nr5r5’表示邻苯二甲酰亚胺、或硝基,
123、x表示卤素原子或离去基团)
124、(2)在碱性条件下将式(v)的r1所示的保护基脱保护的步骤,和
125、(3)根据任意选择,在上述(1)之前,使式(iii)所示的化合物的r4为nhcor3,或在上述(1)之后且上述(2)之前,使式(v)所示的化合物的r4为nhcor3的步骤。
126、上述的(1)的反应中包括偶联反应,上述(3)的反应中包括缩合反应。根据本发明的优选的一个实施方式,在上述(1)之后且上述(2)之前,使式(v)所示的化合物的r4为nhcor3。
127、在本发明的一个实施方式中,提供还包括如下工序的制造方法:上述式(iii)所示的化合物的r4和/或上述式(v)所示的化合物的r4为nr5r5’,并且r5和r5’中的至少一个为氨基的保护基,将上述氨基的保护基脱保护。在本发明的优选的一个方式中,将氨基的保护基在上述(1)之前、或在上述(1)之后并且在上述(2)之前脱保护,优选在上述(1)之后且上述(2)之前脱保护。
128、接着,对在本发明的一个实施方式中的脱保护之前进行的缩合反应和sonogashira coupling(薗头偶联)进行记载。这里所说的“本发明的一个实施方式中的脱保护之前”是指在上述的“末端乙炔的保护基的脱保护”之前,可以应用于马上要该脱保护之前或者在更早。并且,这里所说的“缩合反应”和“薗头偶联”的顺序没有限制,也可以在“缩合反应”和“薗头偶联”之间还有其它的反应工序。
129、(r4为nhcor3时)
130、在上述式(iii)的化合物中r4为nhcor3时,以下述的式(iii-2)所示的化合物为起始物质,进行上述工序(1)和(2),得到式(i)所示的化合物。此时,能够不进行工序(3)而得到式(i)所示的化合物。在下述反应式中,“deprotection”是指“脱保护”。
131、
132、(r4为nr5r5’时)
133、在本发明的一个实施方式中r4为nr5r5’时,以下述的式(iii-1)为起始物质,通过在上述(1)之前使式(iii)所示的化合物的r4为nhcor3、或在上述(1)之后且上述(2)之前使式(v)所示的化合物的r4为nhcor3,在最后在碱性条件下进行脱保护,由此,能够制造式(i)的化合物。
134、
135、以下表示这样的式(i)的化合物的制造步骤(合成路径1)。合成路径1中的“condense”是指“缩合反应”,“deprotection”是指“脱保护”。
136、合成路径1
137、
138、在本发明的一个实施方式中,r5和r5’分别独立地表示氢原子、或氨基的保护基,或者nr5r5’表示邻苯二甲酰亚胺、或硝基。
139、r5和/或r5’为氨基的保护基时,或nr5r5’为邻苯二甲酰亚胺时,从合成路径1中的式(iii-1)所示的化合物向式(iii-2)所示的化合物的衍生、和从式(v-1)所示的化合物向式(ii)所示的化合物的衍生中包括r5和/或r5’的脱保护的工序。
140、r5和r5’中的“保护基”能够列举与r2所列举的保护基相同的保护基,优选为氨基甲酸酯保护基,更优选为boc。
141、在本发明中,r5和r5’分别独立地表示氢原子或保护基,或者nr5r5’表示邻苯二甲酰亚胺或硝基,优选为r5和r5’分别独立地表示氢原子或保护基,更优选为r5和r5’中的至少一个表示保护基,更优选为r5和r5’中的至少一个表示boc,特别优选为r5为氢原子或boc,r5’为氢原子。
142、nr5r5’为硝基时,合成路径1中的从式(iii-1)所示的化合物向式(iii-2)所示的化合物的衍生、和从式(v-1)所示的化合物向式(ii)所示的化合物的衍生中包括将硝基还原为氨基的工序。这样的还原能够采用通常公知的方法,具体而言,能够采用后述的“缩合反应”的项目所述的方法。
143、该合成路径由于避免ohira-bestmann法的使用,能够以任意顺序采用薗头偶联、缩合反应(condense)、和从式(ii)向式(i)的衍生,从能够避免柱色谱法的使用、收率高、并且比专利文献1记载的制造方法降低杂质的含量的方面考虑,优选用以下的(a)~(c)的顺序制造式(i)所示的化合物。
144、(a)通过式(iii-1)所示的化合物与式(viii)所示的化合物的连接而衍生为式(iii-2)所示的化合物,或通过式(iii-1)所示的化合物与式(iv)所示的化合物的连接而衍生为式(v-1)所示的化合物
145、(b)通过式(iii-2)所示的化合物与式(iv)所示的化合物的连接、或式(v-1)所示的化合物与式(viii)所示的化合物的连接而衍生为式(ii)所示的化合物
146、(c)在碱性条件下将式(ii)所示的化合物衍生为式(i)所示的化合物
147、例如,x为溴原子、r1为tes、r2为氨基、r3为5-甲基吡嗪-2-基、nr5r5’为被boc保护的氨基(例如nhboc,即r5为r5’中任一个为氢原子、另一个为氨基的保护基)时,能够从叔丁基(4-(4-氨基-6-溴-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2,2,1]庚烷-1-基)氨基甲酸酯以任意的顺序进行薗头偶联、boc的脱保护(de boc)、和缩合反应(condense)后,通过tes的脱保护(de tes)来制造化合物(1)。
148、
149、在以下表示这样的合成步骤(合成路径2)。
150、合成路径2
151、
152、该合成路径由于避免ohira-bestmann法的使用,能够以任意顺序采用薗头偶联、boc的脱保护(de boc)、缩合反应(condense)、和tes的脱保护(de tes),从能够避免柱色谱法的使用、收率高、并且比专利文献1记载的制造方法降低杂质的含量的方面考虑,优选用以下的(a)~(d)的顺序制造化合物(1)。
153、(a)叔丁基(4-(4-氨基-6-溴-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2,2,1]庚烷-1-基)氨基甲酸酯和三乙基甲硅烷基乙炔的偶联
154、(b)(a)中所得到的化合物的boc的脱保护
155、(c)(b)中所得到的化合物与5-甲基吡嗪-2-羧酸的缩合
156、(d)碱性条件下(c)中所得到的化合物的tes的脱保护
157、更优选为用以下的(a)~(d)的顺序制造化合物(1)。
158、(a)使用钯催化剂和铜催化剂进行的叔丁基(4-(4-氨基-6-溴-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2,2,1]庚烷-1-基)氨基甲酸酯和三乙基甲硅烷基乙炔的偶联
159、(b)(a)所得到的化合物的boc的脱保护
160、(c)使用edc和hobt的组合进行的(b)中所得到的化合物与5-甲基吡嗪-2-羧酸的缩合
161、(d)在利用不含氟化物离子的试剂进行了调整的碱性条件下,对(c)中所得到的化合物的tes的脱保护
162、这样操作而得到式(i)的化合物或其盐时,可以列举式(i-1)所示的杂质的含量少。杂质的含量能够在利用hplc(高速液体色谱)、uplc(超高速高分离液体色谱)的测定中以特定的波长得到的峰面积的面积的比例(%)求出。并且,该波长能够根据化合物的性质适当设定。这对于维持医药品的品质是重要的,考虑人用药品技术要求国际协调理事会(ich)所规定的基准等,式(i-1)所示的杂质的含量可以列举为0.2%以下。优选为0.15%以下,更优选为0.1%以下,更优选为0.05%以下。
163、在本发明的一个实施方式中的制造方法所制造的上述式(i)所示的化合物及其盐中,式(i-1)所示的化合物的含量为0.2%以下,优选为0.15%以下,更优选为0.1%以下,更优选为0.05%以下。此外,式(i-1)所示的化合物也可以不以本发明的一个实施方式中的制造方法制造,即上述比率为检测界限以下即可。
164、
165、(式中,
166、r3表示任选地具有取代基的5~10元的单环式或者多环式的不饱和杂环基)
167、根据本发明的一个方式,能够提供包含式(i)所示的化合物或其盐、以上述的方法所得到的组合物。本发明的一个实施方式中的组合物包含式(i)所示的化合物或其盐、和式(i-1)所示的化合物。在优选的实施方式中,组合物中的式(i-1)所示的化合物的比率为0.2%以下,优选为0.15%以下,更优选为0.1%以下,更优选为0.05%以下。
168、该制造方法中产生的杂质能够使用已知的装置、通过已知的方法检测。例如能够使用hplc检测。hplc能够使用通常市售的装置。
169、在本发明中,式(i-1)所示的化合物或其盐,通过确认在包含化合物(1)或其盐的制剂或api的稳定性、及其制造工序中有无在红外吸收(ir)谱、核磁共振(nmr)谱、hplc、薄层色谱(tlc)等的色谱的测定中的各类似物的存在,能够作为用于管理品质的标准品使用。作为式(i-1)所示的化合物,优选为n-(4-(6-乙炔基-4-(5-甲基吡嗪-2-羧酰胺)-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-羧酰胺。
170、(缩合反应)
171、本发明中进行的缩合反应是用于将下述式(iii-1)或式(v-1)所示的化合物的胺与下述式(viii)所示的羧酸进行缩合而衍生为具有酰胺的式(iii-2)所示的化合物或式(ii)所示的化合物的工序。即,该缩合反应用于构建式(iii-2)所示的化合物或式(ii)所示的化合物的-nhcor3所示的酰胺键。
172、
173、(x表示卤素原子或离去基团,r2、r5和r5’的定义和实施方式与上述相同。)
174、
175、(上述各式中,x表示卤素原子或离去基团,r1、r2、r3、r5和r5’的定义和实施方式与上述相同。)
176、在说明书中,式(iii-1)或式(iii-2)所示的化合物的x为卤素原子或离去基团。作为“卤素原子”,可以列举氟原子、氯原子、溴原子和碘原子,优选为氯原子、溴原子和碘原子,更优选为溴原子和碘原子,特别优选为溴原子。
177、作为“离去基团”,可以列举上述离去基团。优选为为-oso2cnfn+2(n表示1~4的整数)、甲磺酸基、甲苯磺酸基、硝基苯磺酸基和-oso2ph。或者,离去基团优选为-oso2cnf2n+1(n表示1~4的整数)、甲磺酸基、甲苯磺酸基、硝基苯磺酸基和-oso2ph。
178、本缩合反应中所使用的式(iii-1)或式(v-1)所示的化合物中,r5和r5’为氢原子时,能够通过以下所示的方法通过与式(viii)的缩合构建酰胺。
179、在本缩合反应中,相对于式(iii-1)或式(v-1)所示的化合物1摩尔,式(viii)所示的化合物例如为0.5~5.0摩尔,优选为0.7~2.5摩尔,更优选为0.8~1.5摩尔,进一步优选为0.9~1.3摩尔,特别优选为0.9~1.1摩尔。
180、本缩合反应中所使用的式(iii-1)或式(v-1)所示的化合物中,r5和r5’中的至少一个表示氨基的保护基、或nr5r5’表示邻苯二甲酰亚胺时,需要通过适当方法将氨基的保护基脱保护,将r5和r5’衍生为氢原子。
181、作为将r5的保护基的脱保护或将邻苯二甲酰亚胺衍生为氨基的方法,根据各个保护基能够采用通常公知的方法,具体而言,能够采用酸性条件、碱性条件、还原、亲核反应等的方法。在酸性条件下,能够将boc、alloc、邻苯二甲酰亚胺、cbz脱保护,在碱性条件下,能够将fmoc脱保护,在还原条件下,能够将cbz、alloc脱保护。在亲核反应中,能够将邻苯二甲酰亚胺、ns脱保护。在氟化物离子的反应中,能够将ses脱保护。在本发明的r5的脱保护中,优选为酸性条件下的脱保护,更优选为酸性条件下的boc的脱保护。
182、本缩合反应中所使用的式(iii-1)或式(v-1)所示的化合物中,nr5r5’表示硝基时,需要通过适当的方法将r5和r5’衍生为氢原子。
183、该衍生能够采用通常公知的方法,例如,能够采用使用pd、pt、ni等金属和氢源而进行的接触氢化的还原、使用fe、sn、zn等金属和氯化铵、盐酸、乙酸等酸而进行的还原、使用氢化硼钠、氢化锂铝等氢化物而进行的还原。
184、在本发明中,上述的缩合反应能够在后述的乙炔的构建前后的任意时刻进行。即,能够从式(iii-1)和式(v-1)分别衍生为式(iii-2)和式(ii)。该缩合反应优选在末端乙炔的构建后进行。
185、缩合反应中使用的缩合剂只要使反应进行即可,没有特别限制。例如,可以列举:1-(3-二甲基氨基丙基)-3-乙基碳二亚胺或其盐酸盐(wsc或edc)、二环己基碳二亚胺(dcc)、1-环己基-3-(2-吗啉代乙基)碳二亚胺甲基-对甲苯磺酸盐(cmc)、二异丙基碳二亚胺(dic)、1,3-双(2,2-二甲基-1,3-二氧戊烷-4-基甲基)碳二亚胺(bddc)等碳二亚胺缩合剂与1-羟基苯并三唑(hobt)、1-羟基-7-氮杂苯并三唑(hoat)、乙基(羟基亚氨基)氰基乙酸乙酯(oxyma)、n-羟基琥珀酰亚胺(hosu)、五氟苯酚等活性化酯构建用醇的组合、o-(7-氮杂苯并三唑-1-基)-n,n,n’,n’-四甲基四甲基六脲六氟磷酸盐(hatu)、(1-氰基-2-乙氧基-2-氧代亚乙基氨基氧)二甲基氨基吗啉代碳鎓六氟磷酸盐(comu)等脲阳离子缩合剂、2-氯-6-甲基-1,3-二苯基吡啶四氟硼酸盐、1-甲基-2-氯吡啶碘化物等2-卤代-n-烷基吡啶盐(向山缩合试剂)、n,n’-羰基二咪唑(cdi)、1,1’-羰基二(1,2,4-三唑)(cdt)等的咪唑缩合剂、溴三吡咯烷膦六氟磷酸盐、氯三吡咯烷膦六氟磷酸盐、六氟磷酸1h-苯并三唑-1-基氧三(二甲基氨基)膦(bop)、六氟磷酸(苯并三唑-1-基氧)三吡咯烷膦(pybop)、六氟磷酸(7-氮杂苯并三唑-1-基氧)三吡咯烷膦(pyaop)等膦缩合剂、4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉氯化物(dmtmm)、4,6-二甲氧基-1,3,5-三嗪-2-基)-(辛氧基-2-氧代乙基)二甲基三氟甲磺酸铵等三嗪缩合剂、2,4,6-三丙基-1,3,5,2,4,6-三氧三磷烷-2,4,6-三氧化物(t3p)、叠氮化二苯磷(dppa)等。优选为碳二亚胺缩合剂和活性化酯构建用醇的组合,更优选为edc和hobt的组合。此外,这些缩合剂等中包括用聚合物载持的试剂。
186、缩合反应中所使用的溶剂只要使该反应进行即可,没有特别限制。能够选自与硅连接保护基的脱保护工序中所使用的溶剂相同的溶剂。优选为二氯甲烷、氯仿、乙酸乙酯、二甲亚砜、二乙醚、四氢呋喃、1,4-二噁烷、丙酮、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、甲苯、乙腈、四氢呋喃,进一步优选为二氯甲烷、氯仿、乙酸乙酯、二甲亚砜、n,n-二甲基甲酰胺,更进一步优选为二氯甲烷、氯仿。另外,使用的溶剂量只要使反应进行即可,没有特别限制,相对于反应物1kg,优选为5~100l,更优选为15~50l,更进一步优选为25~35l。
187、在末端乙炔的构建后进行本缩合反应时,可以预先将末端乙炔用硅连接保护基进行保护。
188、缩合工序的反应时间只要是能够实现缩合的时间即可,没有特别限制,能够适当设定。例如为1~120小时,优选为2~48小时,进一步优选为5~48小时,优选为8~36小时,进一步优选为14~24小时。
189、缩合工序的反应温度只要是能够实现缩合的温度即可,没有特别限制,能够适当设定。例如为0~60℃,优选为10~50℃,进一步优选为20~40℃。
190、此外,除了与缩合反应中所使用的式(viii)所示的缩合剂的组合以外,可以列举式(viii-1)所示的化合物或者式(viii-2)所示的化合物与碱的组合。
191、
192、其中,式中l表示卤素原子或离去基团,(viii-1)中也包含混合酸酐,定义和实施方式与上述同样,优选为卤素原子,更优选为氯原子。
193、作为与式(viii-1)所示的化合物或者式(viii-2)所示的化合物组合的碱,可以列举三甲胺、三乙胺、二异丙基乙基胺、n-甲基吗啉、二氮杂双环十一烯(dbu)、二氮杂双环壬烯(dbn)、吡啶、4-二甲基氨基吡啶(dmap)等的有机胺;碱金属氢氧化物(氢氧化锂、氢氧化钠、氢氧化钾等)、碱土金属氢氧化物(氢氧化钡、氢氧化钙、乙酸钙等)、碱金属碳酸盐(碳酸氢钠、碳酸钠、碳酸钾、碳酸铯等)、碱土金属碳酸盐(碳酸钡、碳酸钙)、碱金属磷酸盐(磷酸钠、磷酸钾等)、乙酸盐(乙酸镁、乙酸钾、乙酸钙、乙酸铯等)等的无机碱。
194、接着,对末端乙炔的构建进行记载。
195、作为用于构建末端乙炔的方法,一般而言,可以列举corey-fuchs法(コーリー·フックス法)、seyferth-gilbert法(セイファース·ギルバート法)、和ohira-bestmann法(大平·ベストマン法)、薗头偶联(sonogashira coupling)等。corey-fuchs法由于需要醛作为二卤代烯烃的前体,通用性低于需要芳基卤化物的薗头偶联。另外,由于corey-fuchs法需要使用强碱,需要能够耐受强碱条件的基质。另外,seyferth-gilbert法和ohira-bestmann法需要使用具有爆炸性的1-二氮杂-2-氧代丙基膦酸酯,因此,在医药品原料的大量合成方面存在担心。
196、另一方面,薗头偶联是乙炔衍生物与芳基卤化物或炔烃卤化物的交叉偶联反应,具有能够不需要使用强碱或具有爆炸性二氮化合物来进行这样的优点。另外,该乙炔衍生物只要是乙炔所具有的2个氢之中的1个被保护基取代而成的式(iv)所示的化合物即可,能够伴随脱保护而构建通式(i)等所示的末端乙炔。保护基的具体例如r1所示。
197、在本发明中,薗头偶联能够使式(iii)所示的化合物和下述式(iv)所示的化合物反应,合成式(v)的化合物。
198、
199、
200、(上述各式中,x、r1、r2与上述定义相同。r4表示nhcor3或nr5r5’,r5和r5’分别独立地表示氢或氨基的保护基,或者nr5r5’表示邻苯二甲酰亚胺或硝基)
201、通过薗头偶联将式(iv)所示的化合物导入式(iii)所示的化合物中时,式(iii)所示的化合物和式(iv)所示的化合物的量比、该反应的催化剂的配体、溶剂的种类和量、添加顺序对于高纯度化也是重要的因素。
202、在薗头偶联中,相对于式(iii)所示的化合物1摩尔,式(iv)所示的化合物例如为1.0~5.0摩尔,优选为1.25~3.75摩尔,进一步优选为1.5~2.5摩尔。
203、作为薗头偶联的催化剂,只要能够促进反应即可,没有特别限定,例如,能够单独使用钯催化剂、或根据需要组合活化剂来使用。
204、作为钯催化剂,没有特别限制,能够列举碳载持钯(pd-c)、氧化铝载持钯、二氧化硅凝胶固定化钯、乙酸钯、二氯双[二叔丁基(4-二甲基氨基苯基)膦]钯(ii)、四(三苯基膦)钯(0)、二氯(1,1-双(二苯基膦)二茂铁)钯(ii)、二氯双(三苯基膦)钯(ii)等。作为催化剂的配体,没有特别限制,三(邻甲苯基)膦、三(叔丁基)膦、三苯基膦、二氯双(三苯基膦)、2-二叔丁基膦-2’,4’,6’-三异丙基联苯、2-二环己基膦-2’,6’-二甲氧基联苯、[4-(n,n-二甲基氨基)苯基]二叔丁基膦、二叔丁基(4-二甲基氨基苯基)膦、三环己基膦、4,5-双(二苯基膦)-9,9-二甲基黄嘌呤(xantphos)、双(2-(二苯基膦)苯基)醚等。催化剂的配体可以预先在钯催化剂配位的配体,也可以将钯催化剂和催化剂的配体分别加入溶剂中,在反应溶剂中使其配位。
205、钯催化剂如上所述,优选为二氯双[二叔丁基(4-二甲基氨基苯基)膦]钯(ii)。另外,作为其配体,优选为二叔丁基(4-二甲基氨基苯基)膦、三环己基膦。
206、作为活化剂,能够使用铜催化剂、银催化剂、季铵、胺水溶液等。作为铜催化剂,可以列举具有1价铜离子的盐,具体而言,可以列举卤化铜(i)(氟化铜(i)、氯化铜(i)、溴化铜(i)、碘化铜(i)、三氟甲磺酸铜(i)等)。作为银催化剂,可以列举具有1价银离子的盐,具体而言,可以列举氧化银(i)。作为季铵,可以列举氢氧化季铵(氢氧化四正丁基铵:tbaoh;氢氧化四乙基铵:teaoh;正十六烷基三甲基氢氧化铵;氢氧化胆碱等)。作为胺水溶液,可以列举氨、伯胺、仲胺、叔胺等的水溶液。作为活性剂,优选为铜催化剂,更优选为具有1价铜离子的盐,更优选为溴化铜(i)、碘化铜(i)、或三氟甲磺酸铜(i),特别优选为碘化铜(i)。
207、作为薗头偶联的碱,只要是能够实现炔烃的导入的碱即可,没有特别限制,例如,能够列举三甲胺、二乙胺、二异丙胺(dipea)、三乙胺(tea)、n-甲基吗啉、二氮杂双环壬烯(dbn)、吡啶、4-二甲基氨基吡啶(dmap)、吡啶、吗啉、喹啉、哌啶、二氮杂双环十一烯(dbu)等有机碱、或碱金属氢氧化物(氢氧化锂、氢氧化钠、氢氧化钾等)、碱土金属氢氧化物(氢氧化钡、氢氧化钙、乙酸钙等)、碱金属碳酸盐(碳酸氢钠、碳酸钠、碳酸钾、碳酸铯等)、碱土金属碳酸盐(碳酸钡、碳酸钙)、碱金属磷酸盐(磷酸钠、磷酸钾等)、乙酸盐(乙酸镁、乙酸钾、乙酸钙、乙酸铯等)等无机碱。优选为有机碱,更优选为二异丙基乙基胺。
208、作为薗头偶联的溶剂,只要是能够进行该反应的溶剂即可,没有特别限制,例如可以列举n,n-二异丙基乙基胺、n-甲基-2-吡咯烷酮、二甲亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、四氢呋喃、乙酸乙酯、二氯甲烷、氯仿、1,4-二噁烷、甲苯。优选为n-甲基-2-吡咯烷酮、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺。
209、薗头偶联的反应时间只要是能够实现炔烃的导入的时间即可,没有特别限制,能够适当设定。例如为0.5~8小时,优选为0.5~5小时,进一步优选为1~3小时。
210、薗头偶联的反应温度只要是能够实现炔烃的导入的温度即可,没有特别限制,能够适当设定。例如为25~120℃,优选为50~110℃,进一步优选为80~100℃。
211、氨基的保护基的脱保护能够根据保护基通过公知的方法进行脱保护。为酸性条件下的脱保护、还原条件下的脱保护、碱性条件下的脱保护、钯催化剂下的脱保护等。例如,如果是boc,通过在酸性条件下进行脱保护,只要能够进行脱保护,可以使用任意酸设定为酸性条件。优选为三氟乙酸、盐酸、硫酸。另外,用于设定酸性条件的酸的量只要能够进行反应即可,没有特别限制,例如相对于式(i)、式(iii)、式(v)或式(vi)所示的化合物1摩尔,优选为1~100摩尔,更优选为2~50摩尔,更进一步优选3~10摩尔。
212、氨基的保护基的脱保护反应中所使用的溶剂只要能够进行该反应即可,没有特别限制。能够选自与硅连接保护基的脱保护工序中所使用的溶剂相同的溶剂。优选为甲醇、乙醇、2-丙醇、乙腈、四氢呋喃,进一步优选为甲醇、乙醇、2-丙醇,更进一步优选为甲醇。另外,使用的溶剂量只要能够进行反应即可,没有特别限制,相对于反应物1kg,优选为1~100l,更优选为5~50l,更进一步优选为10~30l。
213、通过适当调整上述的碱性条件下的硅连接保护基的脱保护、薗头偶联、向r3的转换、和氨基的保护基的脱保护的顺序、试剂量、溶剂量,即使不利用柱色谱法进行精制也能够制造纯度高的目的化合物。
214、通过上述合成路径制造的化合物(1)可以为非晶质也可以为晶体,为晶体时,可以为单一的晶体,也可以为多种晶体的混晶。晶体化时,能够合成化合物(1)后不作为晶体取出而直接使用,或者暂时作为晶体(粗晶体)取出来使用。另外,能够通过公知的精制方法形成作为目标的单一晶体。另外,本说明书中,晶体可以为水合物或无水物的任一种。
215、化合物(1)或其盐的标记体,即化合物(1)、化合物(1)的盐、将其1个以上的原子用放射性同位元素或者非放射性同位元素取代而得到的化合物也包括在本发明的一个实施方式中。
216、在本说明书中,化学纯度是用hplc测定时的纯度,记为化合物(1)的化学纯度时,是指用hplc测定化合物(1)时的纯度。此时,能够适当设定纯度测定中所使用的检测器的波长。具体而言,化合物(1)的晶体的化学纯度优选为95%以上,更优选为98%以上,特别优选为99%以上。
217、(避免爆炸性试剂的使用)
218、在专利文献1中,通过ohira-bestmann法构建乙炔,制造化合物(1),但是,本发明的制造方法中,不使用ohira-bestmann法。
219、不使用ohira-bestmann法而构建末端乙炔的方法能够使用通常公知的方法。
220、作为其一个实施方式,可以列举:使式(vi)所示的化合物与式(vii)所示的化合物反应,得到式(ix)的化合物;并从式(ix)的化合物得到式(iii-1)所示的化合物。从式(ix)的化合物得到式(iii-1)所示的化合物的方法例如能够使用专利文献1所述的方法。
221、
222、(式中,l表示卤素原子或离去基团)
223、
224、(式(vi)和式(ix)中,
225、r4表示nr5r5’,
226、r5和r5’分别独立地表示氢原子、或氨基的保护基,或者nr5r5’表示邻苯二甲酰亚胺、或硝基,
227、l表示卤素原子或离去基团)
228、(晶体的精制和其它的操作)
229、析出的晶体例如能够通过过滤、水或有机溶剂进行的清洗、加压干燥等公知的分离精制方法,从上述晶体的溶解溶液、混合溶液等分离精制。
230、(活性和用途)
231、通过本发明的一个实施方式的制造方法得到的化合物或其盐或晶体具有优异的egfr抑制活性。其中,对egfr(del19/c797s)、egfr(l858r/c797s)、egfr(del19/t790m/c797s)、egfr(l858r/t790m/c797s)具有优异的抑制活性,作为抗肿瘤剂有用。另外,通过本发明的一个实施方式的制造方法得到的化合物或其盐或晶体具有对于突变型egfr的优异的选择性,具有对野生型egfr、其它激酶的副作用少的优点。
232、本说明书中,“野生型egfr”例如是表示genbank登录号:np_005219.2的氨基酸序列。
233、本说明书中,“exon 19”表示野生型egfr(例如,genbank登录号:np_005219.2)的氨基酸序列中的729-823的区域。
234、本说明书中,“del19”表示野生型egfr的exon 19区域中缺失了1个以上的氨基酸的突变。还包括该区域除了缺失还插入了1个或多个任意的氨基酸的突变。作为exon 19缺失突变,表示exon 19区域的第746位谷氨酸至第750位丙氨酸的5个氨基酸缺失的突变(dele746-a750(或也称为d746-750))、exon 19区域的第747位亮氨酸至第753位脯氨酸的7个氨基酸缺失后插入丝氨酸的突变(del l747-p753inss)、exon 19区域的第747位亮氨酸至第751位苏氨酸的5个氨基酸缺失的突变(del l747-t751)、exon 19区域的第747位亮氨酸至第750位丙氨酸的4个氨基酸缺失后插入脯氨酸的突变(del l747-a750insp)等。优选列举exon 19区域的第746位谷氨酸至第750位丙氨酸的5个氨基酸缺失的突变(del e746-a750)。
235、通过本发明的一个实施方式的制造方法得到的化合物或其盐可以用于将肿瘤以外科方式摘出后为了防止复发而进行的术后辅助化学疗法,也可以用于为了将肿瘤以外科方式摘出而事先进行的术前辅助化学疗法。
236、作为本发明的对象的肿瘤没有特别限制,例如可以列举头颈癌、消化系统癌(食管癌、胃癌、十二指肠癌、肝脏癌、胆道癌(胆囊-胆管癌等)、胰腺癌、大肠癌(结肠直肠癌、结肠癌、直肠癌、肛门癌等)等)、肺癌(非小细胞肺癌、小细胞肺癌、间皮瘤(胸膜间皮瘤、腹膜间皮瘤、心膜间皮瘤、睾丸间皮瘤等))、乳腺癌、生殖系统癌(卵巣癌、外阴癌、子宫癌(宫颈癌、子宫体癌、子宫内膜癌等)等)、泌尿系统癌(肾癌、膀胱癌、前列腺癌、睾丸肿瘤、尿路上皮癌、肾盂癌、尿道癌等)、造血系统肿瘤(白血病、恶性淋巴瘤、多发性骨髓瘤等)、骨-软组织肿瘤、横纹肌肉瘤、皮肤癌、脑肿瘤、恶性神经鞘瘤、神经内分泌肿瘤、甲状腺癌等。优选为头颈癌、乳癌、大肠癌、食管癌、胰腺癌、肺癌、卵巣癌、肾癌、膀胱癌、皮肤癌、脑肿瘤,特别优选为肺癌。此外,这里,癌不仅包含原发灶,还包括转移至其它脏器(肝脏等)的癌。并且,通过本发明的一个实施方式的制造方法得到的化合物或其盐或其晶体具有对于突变型egfr的优异的抑制活性。作为这样的突变型egfr的例子,可以列举药剂耐性突变型egfr、高感受性突变型egfr。因此,本发明的化合物或其盐作为抗肿瘤剂对于具有突变型egfr的上述恶性肿瘤也有用。
237、在本说明书中,化合物的“有效量”的术语是指引起对象的生物学或医学应答、例如酶或蛋白质活性的减少或抑制、或者改善症状、缓解病情、延缓或延迟疾病的进展等的本发明的化合物的量(治疗有效量)。
238、在本说明书中,“对象”的术语包括哺乳动物和非哺乳动物。在一个实施方式中,对象为人,可以为诊断为需要针对本说明书中公开的症状、病情或疾病的处置的人。
239、在使用通过上述制造方法制造的化合物(1)、其盐的晶体或共晶作为医药时,可以将该晶体粉碎或者不粉碎,根据治疗目的采用各种给药形态,能够以通常作为医药品利用的剂型使用。作为该形态,例如可以为片剂、胶囊剂、颗粒剂、细粒剂、散剂、干糖浆剂等口服剂;栓剂、吸入剂、滴鼻剂、软膏剂、贴剂、注射剂等非口服剂的任意剂型。适于这些给药形态的医药组合物能够使用药学上可接受的载体,通过本领域技术人员公知惯用的制剂方法进行制造。
240、作为药学上可接受的载体,可以使用作为制剂原材料惯用的各种有机或无机载体物质,可以作为固形制剂中的赋形剂、结合剂、崩解剂、润滑剂、包衣剂、液态制剂中的溶剂、助溶剂、悬浮剂、等渗剂、缓冲剂、镇痛剂等配合。另外,也可以根据需要使用防腐剂、抗氧化剂、着色剂、甜味剂、稳定剂等制剂添加物。
241、在制备口服用固形制剂的情况下,能够对化合物(1)的晶体(即化合物(1)的游离体的晶体或化合物(1)与酸的晶体(盐的晶体或共晶))加入赋形剂、根据需要加入结合剂、崩解剂、润滑剂、着色剂、矫味和矫味剂等后,通过常规方法制造片剂、包衣片剂、颗粒剂、散剂、胶囊剂等。
242、在制备注射剂的情况下,能够对化合物(1)的晶体(即、化合物(1)的游离体的晶体或化合物(1)与酸的晶体(盐的晶体或共晶))添加ph调节剂-缓冲剂、稳定剂、等渗剂、局部麻醉剂等,通过常规方法制造皮下、肌肉内和静脉内用注射剂。
243、各给药单位形态中应当配合的化合物(1)的晶体(即化合物(1)的游离体的晶体或化合物(1)与酸的晶体(盐的晶体或共晶))的量根据要应用其的患者的症状、或者其剂型等而并不固定,通常而言,在每给药单位形态中,以化合物(1)的游离体换算计,口服剂时希望设为约0.05~1000mg,注射剂时希望设为约0.1~500mg,栓剂或外用剂时希望设为约1~1000mg。
244、另外,具有各给药形态的药剂的上述制造方法所制造的化合物(1)的晶体(即化合物(1)的游离体的晶体或化合物(1)与酸的晶体(盐的晶体或共晶))的每1天的给药量根据患者的症状、体重、年龄、性别等的不同而不同,并不能一概而论,通常成人(体重50kg)每1天,以化合物(1)的游离体换算计,设为约0.05~5000mg、优选为0.1~1000mg即可。
245、(例示的实施方式)
246、以下例示本发明的例示性的实施方式,但是本发明不限定于下述的实施方式。
247、本发明的一个实施方式的制造方法设计用于通过将式(ii)所示的化合物的r1所示的保护基脱保护来制造式(i)所示的化合物或其盐的方法,优选为用于通过在碱性条件下将式(ii)的r1所示的保护基脱保护来制造化合物(1)或其盐的方法,更优选在利用不包含氟化物离子的试剂调整为碱性条件下进行该方法,更优选r1为三乙基甲硅烷基的该方法。
248、另一实施方式中的本发明的制造方法是制造式(i)所示的化合物的方法,其包含以下述(a)~(b)的顺序表示的工序。
249、(a)使式(iv)所示的化合物与式(iii-2)所示的化合物反应,制造式(ii)所示的化合物的工序
250、(b)使式(ii)所示的化合物衍生为式(i)所示的化合物的工序
251、另一实施方式中的本发明的制造方法是式(i)所示的化合物或其盐的制造方法,其包括下述(a)~(c)。
252、(a)通过式(iii-1)所示的化合物与式(viii)所示的化合物的连接而衍生为式(iii-2)所示的化合物、或通过式(iii-1)所示的化合物与式(iv)所示的化合物的连接而制造式(v-1)所示的化合物的步骤;
253、(b)通过式(iii-2)所示的化合物与式(iv)所示的化合物的连接、或通过式(v-1)所示的化合物与式(viii)所示的化合物的连接而衍生为式(ii)所示的化合物的步骤;和
254、(c)通过在碱性条件下将式(ii)的r1所示的保护基脱保护,形成式(i)所示的化合物或其盐的步骤。
255、优选为式(i)所示的化合物或其盐的制造方法,其包括下述(a)~(c)。
256、(a)使式(iv)所示的化合物与式(iii-1)所示的化合物反应,制造式(v-1)所示的化合物的步骤;
257、(b)将式(v-1)所示的化合物衍生为式(ii)所示的化合物的步骤;和
258、(c)在碱性条件下将式(ii)所示的化合物衍生为式(i)所示的化合物的步骤。
259、更优选为式(i)所示的化合物或其盐的制造方法,其中,r1为硅连接保护基,r2为氨基,r3为任选地具有甲基的吡嗪基,r4为nr5r5’,r5为能够在酸性条件下脱保护的保护基,r5’为氢原子,x为卤素原子,l为卤素原子,包括下述(a)~(c),不包括利用柱色谱法的精制工序,式(i-1)所示的化合物的含量为0.2%以下。
260、(a)使式(iv)所示的化合物与式(iii-1)所示的化合物反应,制造式(v-1)所示的化合物的步骤;
261、(b)将式(v-1)所示的化合物的在酸性条件下能够脱保护的保护基脱保护,与式(viii)所示的化合物缩合,衍生为式(ii)所示的化合物的步骤;和
262、(c)在碱性条件下将式(ii)所示的化合物衍生为式(i)所示的化合物的步骤。
263、其中,式(iii-1)所示的化合物是由使式(vi)所示的化合物和式(vii)所示的化合物反应得到的式(ix)的化合物衍生而成的。
264、进一步优选为化合物(1)或其盐的制造方法,其中,r1为硅连接保护基,r2为氨基,r3为5-甲基吡嗪-2-基,r4为nr5r5’,r5为能够在酸性条件下脱保护的保护基,r5’为氢原子,x为卤素原子,l为卤素原子,包括下述(a)~(c),不包括利用柱色谱法的精制工序,化合物(2)的含量为0.2%以下。
265、(a)使式(iv)所示的化合物与式(iii-1)所示的化合物反应,制造式(v-1)所示的化合物的步骤;
266、(b)将式(v-1)所示的化合物的在酸性条件下能够脱保护的保护基脱保护,通过与5-甲基吡嗪-2-羧酸的缩合而衍生为式(ii)所示的化合物的步骤;和
267、(c)在碱性条件下将式(ii)所示的化合物衍生为式(i)所示的化合物的步骤。
268、其中,式(iii-1)所示的化合物从使式(vi)所示的化合物和式(vii)所示的化合物反应得到的式(ix)的化合物衍生而成。
269、更进一步优选为化合物(1)或其盐的制造方法,其中,r1为tes,r2为氨基,r3为5-甲基吡嗪-2-基,r4为nr5r5’,r5为boc,r5’为氢原子,x为溴原子,l为氯原子,包括含下述(a)~(c),不包括利用柱色谱法的精制工序,化合物(2)的含量为0.2%以下。
270、(a)使用钯催化剂和铜催化剂使式(iv)所示的化合物与式(iii-1)所示的化合物反应,制造式(v-1)所示的化合物的步骤;
271、(b)将式(v-1)所示的化合物的boc脱保护,通过与5-甲基吡嗪-2-羧酸的缩合而衍生为式(ii)所示的化合物的步骤;和
272、(c)在利用不包含氟化物离子的试剂进行了调整的碱性条件下将式(ii)所示的化合物衍生为式(i)所示的化合物的步骤。
273、其中,式(iii-1)所示的化合物是由使式(vi)所示的化合物和式(vii)所示的化合物反应而得到的式(ix)的化合物衍生而成的。
274、特别优选为化合物(1)或其盐的制造方法,其中,r1为tes,r2为氨基,r3为5-甲基吡嗪-2-基,r4为nr5r5’,r5为boc,r5’为氢原子,x为溴原子,l表示氯原子,包括下述(a)~(c),不包括利用柱色谱法的精制工序,化合物(2)的含量为0.2%以下。
275、(a)使用二氯双[二叔丁基(4-二甲基氨基苯基)膦]钯(ii)和碘化铜(i)使式(iv)所示的化合物与式(iii-1)所示的化合物反应,制造式(v-1)所示的化合物的工序;
276、(b)将式(v-1)所示的化合物的boc在酸性条件下脱保护,使用edc和hobt的组合与5-甲基吡嗪-2-羧酸进行缩合,由此衍生为式(ii)所示的化合物的工序;
277、(c)将式(ii)所示的化合物在利用碳酸钾进行了调整的碱性条件下衍生为式(i)所示的化合物的工序。
278、其中,式(iii-1)所示的化合物是由使式(vi)所示的化合物和式(vii)所示的化合物反应而得到的式(ix)的化合物衍生而成的。
279、另一实施方式中的本发明的制造方法是制造包含式(i)所示的化合物或其盐和式(i-1)所示的化合物的组合物的方法。该方法中,式(i-1)所示的化合物的比率为0.2%以下,不包括利用柱色谱法的精制工序,包括以下述(a)~(c)的顺序表示的工序。
280、(a)使式(iv)所示的化合物与式(iii-1)所示的化合物反应,制造式(v-1)所示的化合物的工序
281、(b)将式(v-1)所示的化合物衍生为式(ii)所示的化合物的工序
282、(c)在碱性条件下,将式(ii)所示的化合物衍生为式(i)所示的化合物的工序
283、另一实施方式中的本发明的制造方法是制造包含式(i)所示的化合物或其盐和式(i-1)所示的化合物的组合物的方法。该方法中,式(i-1)所示的化合物的比率为0.2%以下,不包括利用柱色谱法的精制工序,包括以下述(a)~(c)的顺序表示的工序。
284、(a)将式(iii-1)所示的化合物衍生为式(iii-2)所示的化合物的工序
285、(b)将式(iii-2)所示的化合物衍生为式(ii)所示的化合物的工序
286、(c)在碱性条件下将式(ii)所示的化合物衍生为式(i)所示的化合物的工序
287、实施例
288、以下,列举实施例更具体的说明本发明,但本发明不受这些例子的任何限定。本发明通过实施例被充分地说明,但可以理解为能够由本领域技术人员进行各种变更和/或修饰。因此,这样的变更和/或修饰只要不脱离本发明的范围,则它们都包括在本发明中。
289、在以下的化合物的实施例中,%只要没有特别说明,表示重量百分比。
290、包括数据处理的装置的操作根据装置所指示的方法和步骤。
291、需要说明的是,从各种谱图获得的数值有时根据测定条件等多少发生变动。因此,这些数值不应被严格解释。
292、装置:waters制uplc(%表示容量百分比)
293、柱:acquity uplc beh c18 column、1.7μm、2.1mm×50mm(waters)
294、柱温:40℃
295、测定波长:254nm
296、流速:0.5ml/分钟
297、流动相a:0.1%甲酸水溶液
298、流动相b:0.1%甲酸-乙腈
299、注射量:1μl
300、梯度:0→0.1分钟:流动相b 5%、0.1→2.1分钟:流动相b 5→95%、2.1→3.5分钟:流动相b 95→98%、3.5→5分钟:流动相b 98→95%。
301、包括数据处理的装置的操作根据装置所指示的方法和步骤。
302、实施例1n-(4-(4-氨基-6-乙炔基-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-甲酰胺(化合物(1))的制造
303、(工序1)叔丁基(4-(4-氨基-5-(喹啉-3-基)-6-((三乙基甲硅烷基)乙炔基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)氨基甲酸酯的合成
304、在通过专利文献1所述的方法合成的叔丁基(4-(4-氨基-6-溴-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)氨基甲酸酯(65.9g)中添加n-甲基吡咯烷酮(1500ml),在氮气环境下以90℃搅拌30分钟使其溶解。冷却至35℃之后,添加二异丙基乙基胺(46.5g)、三乙基甲硅烷基乙炔(33.7g)、碘化铜(i)(4.57g)、二氯双[二叔丁基(4-二甲基氨基苯基)膦]钯(ii)(8.50g)、n-甲基吡咯烷酮(80ml),在氮气环境下以90℃搅拌2小时。冷却至40℃之后,添加10%磷酸二氢钠水溶液、乙酸乙酯。将有机层用稀氨水、水清洗,添加活性炭(33.0g)、sh二氧化硅凝胶(65.9g),在40℃搅拌1小时,在室温搅拌2小时。对不溶物用硅藻土过滤分离之后,将溶剂在减压下蒸馏除去,在所得到的残留物中添加乙腈,在室温下搅拌15小时。过滤获得所析出的固体,用乙腈清洗,并进行减压干燥,由此得到标题化合物(37.4g、收率51%)。
305、(工序2)7-(4-氨基双环[2.2.1]庚烷-1-基)-5-(喹啉-3-基)-6-((三乙基甲硅烷基)乙炔基)-7h-吡咯并[2,3-d]嘧啶-4-胺的合成
306、在如上所述合成的叔丁基(4-(4-氨基-5-(喹啉-3-基)-6-((三乙基甲硅烷基)乙炔基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)氨基甲酸酯(65.0g)中添加5~10%盐酸甲醇溶液(520ml),在氮气环境下以50℃搅拌3小时。在冰冷下,滴加2摩尔/l氢氧化钠水溶液,使ph在12附近。过滤获得所析出的固体,用50%甲醇清洗,并进行减压干燥,由此得到标题化合物(50.3g、收率93%)。
307、(工序3)n-(4-(4-氨基-5-(喹啉-3-基)-6-((三乙基甲硅烷基)乙炔基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-羧酰胺的合成
308、在工序2中得到的7-(4-氨基双环[2.2.1]庚烷-1-基)-5-(喹啉-3-基)-6-((三乙基甲硅烷基)乙炔基)-7h-吡咯并[2,3-d]嘧啶-4-胺(48.3g)、5-甲基吡嗪-2-羧酸(13.4g)、1-羟基苯并三唑1水和物(16.0g)、二异丙基乙基胺(24.6g)和二氯甲烷(1450ml)的混合物中,在氮气环境下添加1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(21.9g),搅拌17小时。将反应液用10%磷酸二氢钠水溶液、饱和碳酸钠水溶液、饱和食盐水清洗,用无水硫酸钠干燥。将干燥剂过滤分离,将溶剂在减压下蒸馏除去,在所得到的残留物中添加甲醇,在室温下搅拌2小时。过滤获得所析出的固体,用甲醇清洗,并进行减压干燥,由此得到标题化合物(54.8g、收率92%)。
309、(工序4)n-(4-(4-氨基-6-乙炔基-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-甲酰胺(化合物1)的合成
310、在工序3所得到的n-(4-(4-氨基-5-(喹啉-3-基)-6-((三乙基甲硅烷基)乙炔基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-羧酰胺(80.0g)、二氯甲烷(1000ml)、甲醇(1000ml)的混合物中,在氮气环境下添加碳酸钾(7.03g)搅拌18小时。将不溶物用硅藻土过滤分离,将溶剂在减压下蒸馏除去,在所得到的残留物中添加甲醇和水,搅拌1小时。过滤得到所析出的固体,用50%甲醇清洗,并进行减压干燥,由此作为淡黄色固体得到标题化合物(64.5g、收率99%)的晶体。
311、对所得到的化合物通过1h-nmr进行分析,结果,得到了与专利文献1的实施例37同样的结果。
312、另外,用uplc进行分析,结果,纯度为99.73%,n-(4-(6-乙炔基-4-(5-甲基吡嗪-2-羧酰胺)-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-羧酰胺的含量为检测极限以下(低于0.05%)。比较例1利用在最终工序具有缩合的方法进行化合物(1)的合成
313、根据专利文献1的实施例37的记载,将专利文献1的制造例1中得到的7-(4-氨基双环[2.2.1]庚烷-1-基)-6-乙炔基-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-4-胺(1当量)、5-甲基吡嗪-2-羧酸(1当量)和二异丙基乙基胺(1.5当量)加入相对于专利文献1的制造例1所得到的化合物1g为20ml的量的二甲亚砜,在其中加入hatu(1.2当量),在室温下搅拌1.5小时。确认反应结束后,加入上述二甲亚砜的约1/3量的水,对所得到的析出物进行过滤并使其干燥,以93%的收率得到化合物(1)。用uplc分析所得到的化合物(1),结果可知,纯度为97.3%,含有1.42%的n-(4-(6-乙炔基-4-(5-甲基吡嗪-2-羧酰胺)-5-(喹啉-3-基)-7h-吡咯并[2,3-d]嘧啶-7-基)双环[2.2.1]庚烷-1-基)-5-甲基吡嗪-2-羧酰胺。
314、需要说明的是,本说明书中记载的全部文献和刊物不论其目的,均通过参照将其整体组合到本说明书中。另外,本说明书包括成为主张本技术的优先权的基础的日本专利申请的特愿2022-071155号(2022年4月22日申请)的专利请求的范围、说明书和附图的公开内容。
315、对本发明的几个实施方式进行了说明,但这些实施方式是作为例子提出的,并不是要限定发明的范围。这些新型的实施方式可以以其它各种方式实施,在不脱离发明的主旨的范围内,能够进行各种省略、置换、变更。这些实施方式或其变形包括在发明的范围和主旨,并且也包括在专利请求的范围所记载的发明及其等同的范围中。
本文地址:https://www.jishuxx.com/zhuanli/20241204/341388.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
上一篇
治疗传染病的新方法与流程
下一篇
返回列表