基于蛋白质图和蛋白质语言模型的核酸结合蛋白识别方法
- 国知局
- 2025-01-10 13:32:48
本发明涉及生物信息学,具体涉及基于蛋白质图和蛋白质语言模型的核酸结合蛋白识别方法。
背景技术:
1、核酸结合蛋白(nabps)在细胞内发挥着多种重要作用。它们通过与dna、rna等核酸相互作用,调节基因表达,维持基因组稳定性,参与信号转导等生物过程。这些蛋白质对细胞的正常功能和体内平衡至关重要。通过对这些蛋白质的精确鉴定和深入研究,我们可以破译它们的细胞功能和它们所处的复杂的调控网络。这一探索不仅揭示了细胞生物学和分子生物学的核心原理,而且增强了我们对基因调控复杂机制的理解。因此,高效准确地鉴定nabps对于理解生物体内复杂的生物反应以及开发新的诊断和治疗方法具有重要意义。
2、在早期阶段,结构比对和统计势被用于鉴定nabps,然而,这些方法受到蛋白质三维(3d)结构数量有限的影响。随着人工智能的兴起,越来越多的机器学习和深度学习算法被用于构建基于序列特征的nabps预测模型。一些研究人员开发了基于序列特征和支持向量机的tripepsvm模型。另一些研究人员利用堆叠算法开发了基于序列特征的stackdppred。还有一些研究人员利用随机森林算法,基于位置特定评分矩阵(position-specific scoring matrix,pssm)和位置特定频率矩阵(position-specific frequencymatrix,psfm)建立了双层模型idrbp-ppct,第一层模型用于区分nabps和非nabps,第二层模型用于区分rna结合蛋白(rbps)、dna结合蛋白(dbps)和dna/rna结合蛋白(drbps)。此外,深度学习算法也被广泛应用于nabps的识别。例如,idrbp_mmc引入了基于卷积神经网络的多标签学习模型来解决交叉预测问题,从而提高了dna结合蛋白和rna结合蛋白的预测性能。论文《idrbp-echf:identifying dna-and rna-binding proteins based onextensible cubic hybrid framework》中提出的idrbp-echf模型是基于五种序列特征和堆叠集成学习算法构建的,使用两种深度学习算法和两种机器学习算法建立基础模型。论文《idrpro-sc:identifying dna-binding proteins andrna-binding proteins basedon subfunction classifiers》中提出的idrpro-sc也是使用基于pssm特征的堆叠集成学习算法构建的,其中使用bilstm构建基本模型来区分nabps的子函数。
3、上述现有技术存在以下问题:
4、结构信息利用不足:由于nabps的3d结构信息的稀缺,现有模型很少利用这些结构信息。然而,结构信息在理解蛋白质与核酸的相互作用中具有重要作用。当前的基于序列的模型,如tripepsvm、stackdppred和idrbp_mmc,主要依赖序列特征进行预测,而未能充分利用3d结构信息,这限制了模型的预测能力和预测准确性。
5、数据覆盖率低:如idrpro-sc所述,具有可用结构信息的蛋白质仅占uniprotkb中所有蛋白质的0.36%。这种低覆盖率限制了模型的广泛应用。许多现有模型(如idrbp-ppct和idrpro-sc)主要依赖少量的3d结构数据,这使得这些模型难以泛化到更广泛的数据集中。
6、预测性能受限:由于未能充分利用3d结构信息,现有模型的预测性能在某些情况下受到限制,尤其是在面对结构复杂的蛋白质时。比如,基于序列特征的stackdppred和基于堆叠集成学习的idrbp-echf和idrpro-sc等模型在处理结构复杂的蛋白质时,无法充分捕捉蛋白质的空间信息,从而影响了预测的准确性和性能。
7、上述问题亟待解决,为此,本发明提出了基于蛋白质图和蛋白质语言模型的核酸结合蛋白识别方法。
技术实现思路
1、本发明所要解决的技术问题在于:如何解决现有技术中存在的结构信息利用不足、数据覆盖率低、预测性能受限等问题,提供了基于蛋白质图和蛋白质语言模型的核酸结合蛋白识别方法。
2、本发明是通过以下技术方案解决上述技术问题的,本发明包括以下步骤:
3、s1:数据集处理
4、从uniprot数据库中检索蛋白质序列数据,并从alphafold数据库中获取相应蛋白质的预测3d结构信息,将序列相似度超过设定值的蛋白质和无法获取结构信息的蛋白质去除,得到训练数据集,然后选定测试数据集;
5、s2:特征提取
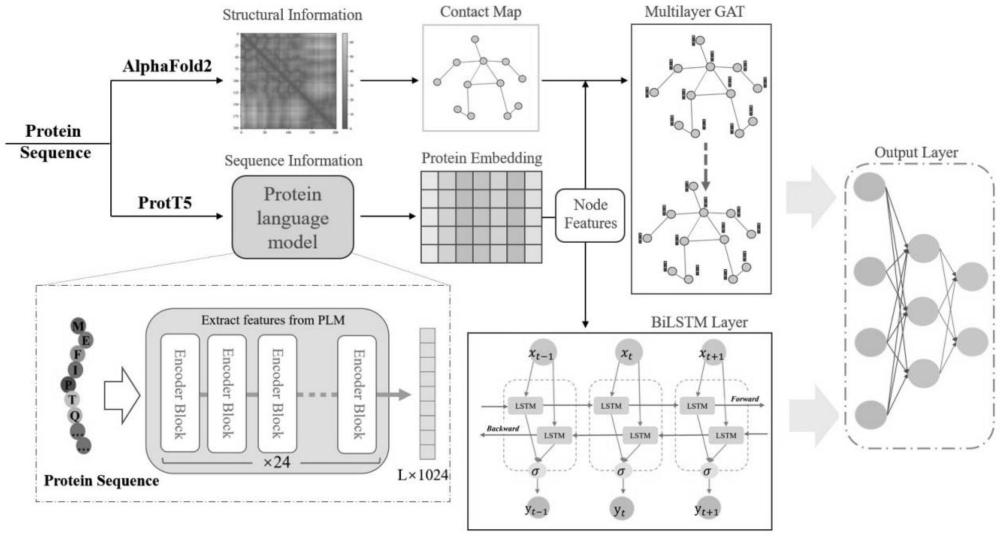
6、使用prott5模型生成蛋白质序列的嵌入特征,并根据预测3d结构信息将蛋白质表示为图,得到蛋白质图,捕捉其结构和序列特征;
7、s3:模型建立
8、利用gat网络、bilstm网络与mlp建立预测模型,在预测模型中,通过gat网络和bilstm网络增强特征表示,然后将gat网络和bilstm网络的输出特征连接起来,通过mlp进行nabps分类,实现对nabps的识别;
9、s4:模型训练
10、利用训练数据集对预测模型进行训练,得到训练后的预测模型;
11、s5:nabps识别
12、利用训练后的预测模型对测试数据集中样本进行nabps识别工作,得到识别结果。
13、更进一步地,在所述步骤s1中,具体处理过程如下:
14、s11:从uniprot数据库搜索具有dna结合和rna结合功能的蛋白质序列,并检索其相应的注释信息;
15、s12:从alphafold数据库中检索到相应蛋白质序列的预测3d结构;
16、s13:使用blastclust工具去除序列相似度超过设定值的蛋白质序列,并去除缺乏预测3d结构信息的蛋白质序列,得到训练数据集;
17、s14:选定数据集pdb255和test474作为测试数据集。
18、更进一步地,在所述步骤s2中,具体处理过程如下:
19、s21:利用prott5模型进行蛋白质语言模型嵌入,从蛋白质序列中提取每个残基的嵌入特征,生成一个大小为l×1024的特征矩阵,其中l为序列长度;
20、s22:通过alphafold数据库进行检索,获取蛋白质的预测3d结构,并将每个蛋白质表示为一个图,每个残基作为节点,节点间的边根据其欧氏距离确定,从而完成蛋白质图的构建工作。
21、更进一步地,在所述步骤s22中,每个蛋白质表示为一个无向图g=(v,h,e,a),其中,v为图g中节点的集合,vi∈v,vi表示第i个残差节点,v为蛋白质中所有残差节点的集合,h为所有节点特征的集合,h∈h|v|×d,|v|是节点的总数,d是节点特征的维数,e是图g中所有边的集合,eij∈e表示节点i和节点j之间的边,a为图g的邻接矩阵,如果任意两个节点之间的欧氏距离小于等于则认为这两个节点是连通的,否则认为这两个节点是不连通的。
22、更进一步地,在所述步骤s3中,gat网络利用注意力机制来获取相邻节点的不同权值,然后将加权相邻节点的信息聚合起来更新节点嵌入;在预测模型中,使用堆叠的多个gat层来实现节点的高级表示,每个gat层通过学习节点与相邻节点之间的关系来动态调整节点特征,在整个gat网络的最后一层,对所有节点的特征进行平均池化,得到整个蛋白质的特征表示,即图嵌入表示。
23、更进一步地,在所述步骤s3中,bilstm网络能够从两个方向捕获序列内部的关系,以保持序列语义的完整性,由prott5模型生成的嵌入特征在lstm内进行处理,通过遗忘门决定从细胞状态中丢弃哪些信息,而新信息则通过输入门和细胞状态更新被合并,新信息包括嵌入特征和前一个时间步的数据。
24、更进一步地,在所述步骤s3中,mlp的处理公式如下:
25、
26、其中,g′表示gat网络的输出,hbi表示bilstm网络的输出,concat表示concat函数,用于将gat网络和bilstm网络的输出特征连接起来,softmax表示softmax函数,用于将结果映射到0到1之间的范围,表示预测结果。
27、本发明相比现有技术具有以下优点:
28、1、将prott5蛋白质语言模型提取的高维序列特征与alphafold预测的三维结构信息结合起来,使得模型能够同时捕捉序列和结构的丰富特征信息,显著提升nabps预测的准确性和可靠性;
29、2、通过将蛋白质表示为图结构,并引入图注意神经网络来处理这些图,gat网络通过注意力机制为每个节点分配不同的权重,整合节点的结构信息;同时,利用bilstm网络处理从prott5模型提取的序列嵌入特征,捕捉序列中的长距离依赖关系,gat网络和bilstm网络均增强了序列特征的表示能力,使得所构建的预测模型能够更全面地理解蛋白质序列中的重要信息,从而提高nabps的预测性能;
30、3、gat网络和bilstm网络输出的特征进行融合,并通过mlp进行nabps的分类,使得预测模型能够充分利用不同来源的特征信息,提高了nabps预测的性能和鲁棒性。
本文地址:https://www.jishuxx.com/zhuanli/20250110/353836.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。