一种Elafibranor的合成方法与流程
- 国知局
- 2025-01-10 13:35:33
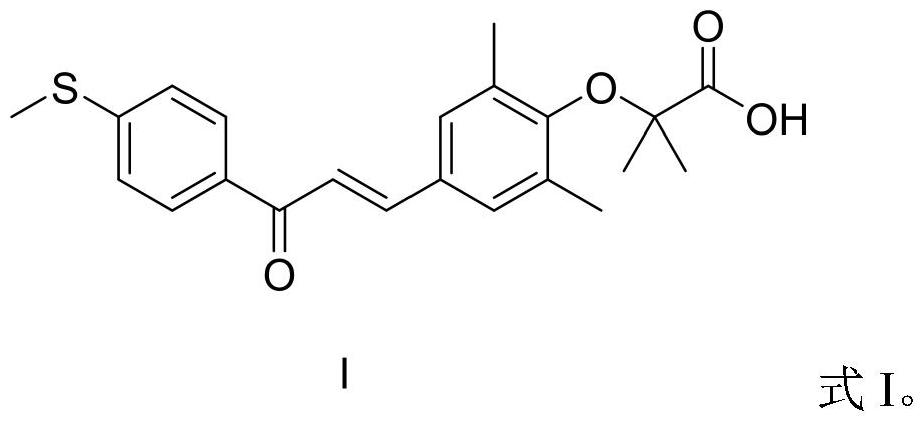
本发明涉及生物医药,具体地,涉及一种elafibranor的合成方法。
背景技术:
1、2024年6月,美国fda加速批准益普生(ipsen)公司的iqirvo(elafibranor)80毫克片剂与熊去氧胆酸(udca)联合用于治疗对udca应答不足的成人原发性胆汁性胆管炎(pbc),或作为单药疗法治疗对udca不耐受的患者。该疗法的获批主要基于关键性3期试验elative的积极结果。分析显示,主要复合终点达到显著治疗获益。分析显示,80mgelafibranor组(51%)达到生化应答的患者与安慰剂组(4%)相比的差异为47%(p<0.001)。在试验中,生化应答定义为第52周时碱性磷酸酶(alp)<1.67×正常值上限(uln),alp降低≥15%,总胆红素(tb)≤uln。alp和胆红素是pbc疾病进展的重要预测因素。两者水平降低可能显示胆汁淤积性损伤减轻,肝功能改善。此外,只有接受elafibranor治疗的患者在第52周达到alp正常值,包含15%药物组患者(p=0.002),这是试验的一个关键次要终点。elafibranor显著的生化效应亦显示于患者alp水平较基线快速降低。
2、elafibranor组患者早在第4周就可观察到alp水平下降,并持续至第52周,与安慰剂组相比,elafibranor组的alp降低幅度达41%。
3、pbc是一种严重的慢性肝病,患者由于胆管的慢性损伤,导致肝脏清除体内毒素的能力下降,引发肝硬化和肝功能衰竭。目前大约有50%的pbc患者对已有疗法应答不良或者无法耐受疗法的毒副作用。这一领域仍然有很大的未竟需求。iqirvo是近十年来首个获批用于治疗罕见肝病原发性胆汁性胆管炎的新药。
4、elafibranor的化学结构式如式i所示:
5、
6、现有技术如us20060142611或us20050176808的合成方法都是由4-甲硫基苯乙酮和3,5-二甲基-4-羟基苯甲醛缩合得到elafibranor的中间体iv,再与2-溴代异丁酸叔丁酯反应得到中间体ⅱ,然后再得到elafibranor。
7、
8、其中,在4-甲硫基苯乙酮和3,5-二甲基-4-羟基苯甲醛缩合过程中,形成的双键的过程中,难以避免产生中间体iv的z式、e式混合物,再和2-溴代异丁酸叔丁酯反应得到的中间体ii,亦为z式、e式混合物。e式异构体极性与z式非常接近,难以结晶纯化,产品需要非常精密的柱层析,不利于工业化生产。
9、因此,目前针对elafibranor的合成方法仍有待改进。
技术实现思路
1、本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一个目的在于提出一种式i所示化合物elafibranor的合成方法。相对于现有技术,本发明所述的方法选用式1所示化合物与三苯基膦构建反应的中间体式2所示化合物,再与式3所示化合物发生缩合反应得到式4所示化合物,式4所示化合物为e式构型,最后式4所示化合物与式5所示化合物发生烷基化反应得到产物elafibranor。
2、在本发明的一个方面,本发明提供了一种式i所示化合物elafibranor的合成方法。根据本发明的实施例,该合成工艺包括:
3、(1)使式1所示化合物与三苯基膦接触,以便获得式2所示化合物;
4、(2)使式2所示化合物与式3所示化合物接触,以便获得式4所示化合物;
5、(3)使式4所示化合物与式5所示化合物、cs2co3、cui接触,以便获得式i所示化合物elafibranor,
6、
7、发明人发现,利用本发明所述的合成方法,以式1所示化合物和三苯基膦为起始原料,其总共经过3步反应,可顺利地合成制备得到目标产物elafibranor。
8、在本文中所使用的术语“接触”应做广义理解,其可以是任何能够使得至少两种反应物发生化学反应的方式,例如可以是将两种反应物在适当的条件下进行混合。根据需要,可以在搅拌下,将需要进行接触的反应物进行混合,由此,搅拌的类型并不受特别限制,例如可以为机械搅拌,即在机械力的作用下进行搅拌。
9、在本文中,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。在本发明的描述中,“多个”的含义是两个或两个以上,除非另有明确具体的限定。
10、根据本发明的实施例,上述制备式2所示化合物、式4所示化合物、式i所示化合物的方法还可以具有下列附加技术特征至少之一:
11、根据本发明的实施例,本发明所述的化学反应可以按照本领域已知的任何方法进行。式2所示化合物、式4所示化合物、式i所示化合物的原料的来源并不受特别限制,其可以是采用任何已知的方法制备的,或者市售获得的。例如式1所示化合物的cas为:42445-46-5,式2所示化合物的cas为:603-35-0,式3所示化合物的cas为:400822-47-1。
12、根据本发明的实施例,在步骤(1)中,式1所示化合物与三苯基膦的接触方式并不受特别限制。由此,可以提升式1所示化合物与三苯基膦接触反应的效率,加快反应速度,进一步提高利用该方法制备式2所示化合物的效率。
13、根据本发明的实施例,在步骤(1)中,包括如下步骤:室温下,将三苯基膦溶于苯中,再加入式1所示化合物,保持室温继续搅拌6~7小时,通过薄层色谱(tlc)监测式1所示化合物完全反应,过滤反应液,用适量苯洗涤滤饼,减压烘干,得到式2所示化合物。由此,可以提升式1所示化合物与三苯基膦接触反应的效率,加快反应速度,进一步提高利用该方法制备式2所示化合物的效率。
14、根据本发明的实施例,在步骤(1)中,式1所示化合物与三苯基膦的摩尔比为1:(1.0~1.2),优选式1所示化合物与三苯基膦的摩尔比为1:1.03。由此,可以进一步提高利用该方法制备式2所示化合物的效率。
15、根据本发明的实施例,在步骤(1)中,优选保持室温继续搅拌6.5小时。
16、根据本发明的一个具体实施例,在步骤(1)中,包括如下步骤:室温下,将三苯基膦(11.02g,42.01mmol)溶于苯(120ml)中,再加入式1所示化合物(10.0g,40.79mmol),保持室温继续搅拌6.5小时,通过薄层色谱(tlc)监测式1所示化合物完全反应,过滤反应液,用适量苯洗涤滤饼,减压烘干,得到式2所示化合物,得量17.26g,收率99.2%。
17、根据本发明的实施例,在步骤(2)中,式2所示化合物与式3所示化合物的接触方式并不受特别限制。由此,可以提升式2所示化合物与式3所示化合物接触反应的效率,加快反应速度,进一步提高利用该方法制备式4所示化合物的效率。
18、根据本发明的实施例,在步骤(2)中,包括如下步骤:室温下,将式2所示化合物和式3所示化合物溶于无水四氢呋喃中,保持在室温下,将反应液搅拌反应45~55小时,反应毕,将反应液减压浓缩后,浓缩物用石油醚/乙酸乙酯混合溶剂,经硅胶柱层析纯化,得到式4所示化合物。由此,可以提升式2所示化合物与式3所示化合物接触反应的效率,加快反应速度,进一步提高利用该方法制备式4所示化合物的效率。
19、根据本发明的实施例,在步骤(2)中,式2所示化合物与式3所示化合物的摩尔比为(2.5~4):1,优选式2所示化合物与式3所示化合物的摩尔比为3:1。由此,可以进一步提高利用该方法制备式4所示化合物的效率。
20、根据本发明的实施例,在步骤(2)中,石油醚/乙酸乙酯混合溶剂中石油醚与乙酸乙酯的体积比为(6~12):1,优选石油醚/乙酸乙酯混合溶剂的体积比为10:1。
21、根据本发明的实施例,在步骤(2)中,优选反应液搅拌反应48小时。
22、根据本发明的一个具体实施例,在步骤(2)中,包括如下步骤:室温下,将式2所示化合物(15.0g,35.17mmol)和式3所示化合物(2.5g,11.73mmol)溶于无水四氢呋喃(150ml)中,保持在室温下,将反应液搅拌反应48小时,反应毕,将反应液减压浓缩后,浓缩物用体积比为10:1的石油醚/乙酸乙酯混合溶剂,经硅胶柱层析纯化,得到式4所示化合物,得量4.10g,收率96.7%。
23、lc-ms(apci):m/z=361.0(m+1)+。
24、1h nmr(400mhz,cdcl3)δ7.99(d,j=8.6hz,1h);7.75(d,j=15.4hz,2h);7.42(d,j=15.5hz,1h);7.36-7.28(m,4h);2.57(s,3h);2.32(s,6h).
25、经scifinder检索,式4所示化合物为未见文献报道的新化合物。
26、根据本发明的具体实施例,式4所示化合物的用途为:作为制备式i所示化合物elafibranor的中间体化合物。
27、根据本发明的实施例,在步骤(3)中,式4所示化合物与式5所示化合物、cs2co3、cui的接触方式并不受特别限制。由此,可以提升式4所示化合物与式5所示化合物、cs2co3、cui接触反应的效率,加快反应速度,进一步提高利用该方法制备式i所示化合物的效率。
28、根据本发明的实施例,在步骤(3)中,包括如下步骤:氮气气氛下,向dmf中加入式4所示化合物和式5所示化合物,搅拌,加入cs2co3、碘化亚铜(cui),反应液升至90~110℃后搅拌10~13小时,tlc显示式4所示化合物完全反应,将反应液用硅藻土过滤,滤液减压浓缩,尽量除去dmf,剩余液体加入乙酸乙酯溶解,再加入hcl溶液洗涤,有机相减压浓缩,加入石油醚/乙酸乙酯混合溶剂重结晶,过滤,减压干燥,得到式i所示化合物elafibranor。由此,可以提升式4所示化合物与式5所示化合物、cs2co3、cui接触反应的效率,加快反应速度,进一步提高利用该方法制备式i所示化合物的效率。
29、根据本发明的实施例,在步骤(3)中,式4所示化合物与式5所示化合物、cs2co3、cui的摩尔比为1:(1.1~1.6):(1.2~2.5):(0.15~0.4),优选式4所示化合物与式5所示化合物、cs2co3、cui的摩尔比为1:1.3:1.5:0.2。由此,可以进一步提高利用该方法制备式i所示化合物的效率。
30、根据本发明的实施例,在步骤(3)中,所述石油醚/乙酸乙酯混合溶剂中,石油醚与乙酸乙酯的体积比为10:1。
31、根据本发明的实施例,在步骤(3)中,优选反应液升至98~100℃后搅拌10小时。
32、根据本发明的一个具体实施例,在步骤(3)中,包括如下步骤:氮气气氛下,向dmf(400ml)中加入式4所示化合物(35.0g,96.87mmol)和式5所示化合物(13.1g,125.9mmol),搅拌,加入cs2co3(47.34g,145.3mmol)、碘化亚铜(cui)(3.70g,19.4mmol),反应液升至98~100℃后搅拌10小时,tlc显示式4所示化合物完全反应,将反应液用硅藻土过滤,滤液减压浓缩,尽量除去dmf,剩余液体加入500ml乙酸乙酯溶解,再加入1m的hcl溶液(约300ml)洗涤,有机相减压浓缩,加入300ml石油醚/乙酸乙酯混合溶剂(体积比为10:1)重结晶,过滤,减压干燥,得到式i所示化合物elafibranor,得量31.47g,收率84.5%,纯度99.7%(hplc)。
33、根据本发明的具体实施例,式i所示化合物elafibranor的合成路线可以如下所示:
34、
35、相对于现有技术,本发明所述的elafibranor的合成方法,其至少具有以下有益效果:
36、1、相对于现有技术,本发明所述的方法选用式1所示化合物与三苯基膦构建反应的中间体中间体式2所示化合物,再与式3所示化合物发生缩合反应得到式4所示化合物,式4所示化合物为e式构型,最后式4所示化合物与式5所示化合物发生烷基化反应得到产物elafibranor。
37、2、现有技术在4-甲硫基苯乙酮和3,5-二甲基-4-羟基苯甲醛缩合过程中,形成的双键的过程中,难以避免产生中间体iv的z式、e式混合物,再和2-溴代异丁酸叔丁酯反应得到的中间体ii,亦为z式、e式混合物。e式异构体极性与z式非常接近,难以结晶纯化,产品需要非常精密的柱层析,不利于工业化生产。本发明采用三苯基膦构建缩合过程的中间体化合物2,再与化合物3生成e式构型。此外,利用三苯基膦中间体可以有效提高缩合过程中的产率,适合工业化生产并具有良好的成本效益。总之本发明所述的合成方法,其合成路线具有收率高、纯化简便等优点,是易于实现产业化,非常适用于工业化大生产的方法。
本文地址:https://www.jishuxx.com/zhuanli/20250110/354085.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表