一种恩塞芬汀的制备方法与流程
- 国知局
- 2025-01-10 13:33:32
本发明涉及生物医药,具体地,涉及一种恩塞芬汀的制备方法。
背景技术:
1、2024年6月26日,美国fda批准veronapharma公司的ohtuvayre(ensifentrine,中文名:恩塞芬汀)上市,用于成人慢性阻塞性肺病(copd)的维持治疗。恩塞芬汀是20多年来首个具有新颖作用机制的吸入性产品,为copd患者提供了新的治疗选择。恩塞芬汀是一款由verona开发的靶向磷酸二酯酶-3(pde3)和磷酸二酯酶-4(pde4)的双靶点抑制剂,对pde3的亲和度是pde4的3440倍。作为双重pde3/4抑制剂,恩塞芬汀能够提供支气管扩张和抗炎双重作用,显著改善患者的呼吸功能和生活质量;除此以外,恩塞芬汀作为雾化吸入制剂,有助于避免胃肠道相关副作用。作为copd领域全新机制的药物,恩塞芬汀药物总体安全性良好,安慰剂组因副作用停药的患者更多,恩塞芬汀比安慰剂组更多的副作用包括高血压、背痛、鼻咽炎等。
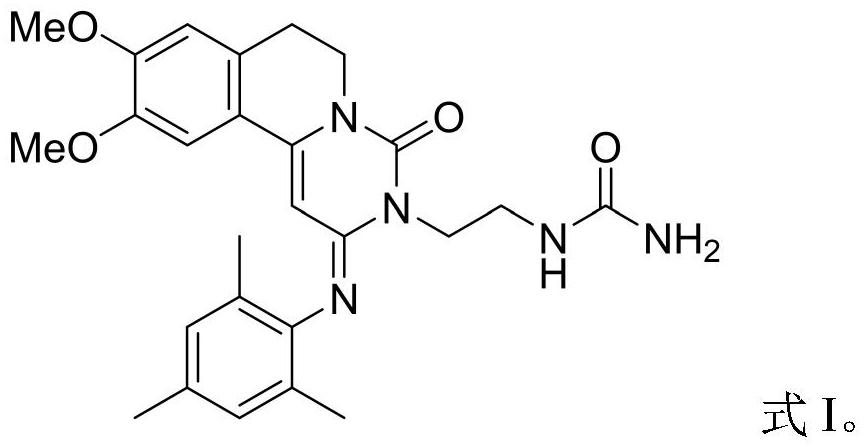
2、恩塞芬汀的化学结构式如式i所示:
3、
4、然而,目前针对恩塞芬汀的制备方法仍有待改进。
技术实现思路
1、本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一个目的在于提出一种式i所示化合物恩塞芬汀的制备方法。相对于现有技术,本发明所述的制备方法,其将初始原料式1所示化合物和式2所示化合物经过sizuki偶联反应得到式3所示化合物,经过分子内成环得到式4所示化合物,利用pocl3将羰基转化为卤化物(式5所示化合物),再与式6所示化合物经过烷基化反应得到式7所示化合物,其中式7所示化合物的酰基亚胺再与式8所示化合物发生取代反应得到产物恩塞芬汀。
2、在本发明的一个方面,本发明提供了一种式i所示化合物恩塞芬汀的制备方法。根据本发明的实施例,该制备方法包括:
3、(1)使式1所示化合物与式2所示化合物、碳酸钾、四(三苯基磷)钯接触,以便获得式3所示化合物;
4、(2)使式3所示化合物与三氟乙酸酐(tfaa)接触,以便获得式4所示化合物;
5、(3)使式4所示化合物与三氯氧磷接触,以便获得式5所示化合物;
6、(4)使式5所示化合物与式6所示化合物接触,以便获得式7所示化合物;
7、(5)使式7所示化合物与式8所示化合物、磷酸钾、碘化物接触,以便获得式i所示化合物恩塞芬汀,
8、
9、其中,所述碘化物为选自碘化钠、或碘化钾的至少一种。
10、发明人发现,利用本发明所述的制备方法,以式1所示化合物和式2所示化合物、碳酸钾、四(三苯基磷)钯为起始原料,其总共经过5步反应,可顺利地合成制备得到目标产物恩塞芬汀。
11、在本文中所使用的术语“接触”应做广义理解,其可以是任何能够使得至少两种反应物发生化学反应的方式,例如可以是将两种反应物在适当的条件下进行混合。根据需要,可以在搅拌下,将需要进行接触的反应物进行混合,由此,搅拌的类型并不受特别限制,例如可以为机械搅拌,即在机械力的作用下进行搅拌。
12、在本文中,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。在本发明的描述中,“多个”的含义是两个或两个以上,除非另有明确具体的限定。
13、根据本发明的实施例,上述制备式3所示化合物、式4所示化合物、式5所示化合物、式7所示化合物、式i所示化合物的方法还可以具有下列附加技术特征至少之一:
14、根据本发明的实施例,本发明所述的化学反应可以按照本领域已知的任何方法进行。式3所示化合物、式4所示化合物、式5所示化合物、式7所示化合物、式i所示化合物的原料的来源并不受特别限制,其可以是采用任何已知的方法制备的,或者市售获得的。例如式1所示化合物的cas为:681431-18-5。
15、根据本发明的实施例,在步骤(1)中,式1所示化合物与式2所示化合物、碳酸钾、四(三苯基磷)钯的接触方式并不受特别限制。由此,可以提升式1所示化合物与式2所示化合物、碳酸钾、四(三苯基磷)钯接触反应的效率,加快反应速度,进一步提高利用该方法制备式3所示化合物的效率。
16、根据本发明的实施例,在步骤(1)中,包括如下步骤:室温下,将式1所示化合物、式2所示化合物、碳酸钾、和四(三苯基磷)钯加入到无水二氧六环中,置换氮气并于95℃~101℃加热反应3小时45分钟~4小时15分钟,反应液降至室温后,用硅藻土过滤,加入水洗涤,再加入乙酸乙酯萃取,有机相加入饱和盐水洗涤,无水硫酸钠干燥,滤液减压浓缩蒸出溶剂,浓缩物用二氯甲烷/甲醇混合溶剂,经硅胶柱层析纯化,得式3所示化合物。由此,可以提升式1所示化合物与式2所示化合物、碳酸钾、四(三苯基磷)钯接触反应的效率,加快反应速度,进一步提高利用该方法制备式3所示化合物的效率。
17、根据本发明的实施例,在步骤(1)中,式1所示化合物与式2所示化合物、碳酸钾、四(三苯基磷)钯的摩尔比为1:(1.0~1.1):(2.5~4.5):(0.05~0.15),优选式1所示化合物与式2所示化合物、碳酸钾、四(三苯基磷)钯的摩尔比为1:1.02:3.0:0.1。由此,可以进一步提高利用该方法制备式3所示化合物的效率。
18、根据本发明的实施例,在步骤(1)中,二氯甲烷/甲醇混合溶剂中二氯甲烷/甲醇的体积比为(8~12):1,优选二氯甲烷/甲醇混合溶剂的体积比为10:1。
19、根据本发明的实施例,在步骤(1)中,优选于98℃~101℃加热4小时。
20、根据本发明的一个具体实施例,在步骤(1)中,包括如下步骤:室温下,将式1所示化合物(22.60g,0.1mol),式2所示化合物(19.48g,0.102mol),碳酸钾(41.46g,0.3mol)和四(三苯基磷)钯(11.56g,0.01mol)加入到无水二氧六环(260ml)中,置换氮气并于98℃~102℃加热反应4小时,反应液降至室温后,用硅藻土过滤,加入200ml水洗涤,再加入200ml乙酸乙酯萃取,有机相加入200ml饱和盐水洗涤,无水硫酸钠干燥,滤液减压浓缩蒸出溶剂,浓缩物用体积比为10:1的二氯甲烷/甲醇混合溶剂,经硅胶柱层析纯化,得到式3所示化合物,得量25.08g,收率85.8%。
21、根据本发明的实施例,在步骤(2)中,式3所示化合物与三氟乙酸酐的接触方式并不受特别限制。由此,可以提升式3所示化合物与三氟乙酸酐接触反应的效率,加快反应速度,进一步提高利用该方法制备式4所示化合物的效率。
22、根据本发明的实施例,在步骤(2)中,包括如下步骤:0℃下,将三氟乙酸酐(tfaa)加入到含式3所示化合物的二氯甲烷溶液中,在室温下搅拌反应0.5小时,反应毕,进行后处理,反应液在0℃下加入冰水淬灭,分离有机层后加入饱和碳酸氢钠溶液洗涤,有机相分离后加入饱和盐水洗涤,经过无水硫酸钠干燥,滤液减压浓缩蒸出溶剂,浓缩物用体积比为3:1的石油醚/乙酸乙酯混合溶剂,经硅胶柱层析纯化,得到式4所示化合物。由此,可以提升式3所示化合物与三氟乙酸酐接触反应的效率,加快反应速度,进一步提高利用该方法制备式4所示化合物的效率。
23、根据本发明的实施例,在步骤(2)中,式3所示化合物与三氟乙酸酐的摩尔比为1:(1.3~2.0),优选式3所示化合物与三氟乙酸酐的摩尔比为1:1.5。由此,可以进一步提高利用该方法制备式4所示化合物的效率。
24、根据本发明的一个具体实施例,在步骤(2)中,包括如下步骤:0℃下,将三氟乙酸酐(25.87g,123.17mmol)加入到含式3所示化合物(24.0g,82.11mmol)的二氯甲烷溶液(240ml)中,在室温下搅拌反应0.5小时,反应毕,进行后处理,反应液在0℃下加入冰水(50ml)淬灭,分离有机层后加入200ml饱和碳酸氢钠溶液洗涤,有机相分离后加入100ml饱和盐水洗涤,经过无水硫酸钠干燥,滤液减压浓缩蒸出溶剂,浓缩物用体积比为3:1的石油醚/乙酸乙酯混合溶剂,经硅胶柱层析纯化,得到式4所示化合物,得量20.29g,收率90.1%。
25、根据本发明的实施例,在步骤(3)中,式4所示化合物与三氯氧磷的接触方式并不受特别限制。由此,可以提升式4所示化合物与三氯氧磷接触反应的效率,加快反应速度,进一步提高利用该方法制备式5所示化合物的效率。
26、根据本发明的实施例,在步骤(3)中,包括如下步骤:室温下,向三氯氧磷中加入式4所示化合物,搅拌升温至回流状态反应3小时,反应液降至室温后,减压浓缩,剩余少量混合物时,加入适量冰水,快速搅拌,滴加饱和naoh溶液至ph=10,将固体过滤,滤饼用冰水洗涤,干燥得到式5所示化合物。由此,可以提升式4所示化合物与三氯氧磷接触反应的效率,加快反应速度,进一步提高利用该方法制备式5所示化合物的效率。
27、根据本发明的实施例,在步骤(3)中,式4所示化合物与三氯氧磷的重量比为1:(5~10),优选式4所示化合物与三氯氧磷的重量比为1:6.3。由此,可以进一步提高利用该方法制备式5所示化合物的效率。
28、根据本发明的一个具体实施例,在步骤(3)中,包括如下步骤:室温下,向三氯氧磷(120ml)中加入式4所示化合物(19.0g,69.27mmol),搅拌升温至回流状态反应3小时,反应液降至室温后,减压浓缩,剩余少量混合物(约30ml)时,加入适量冰水,快速搅拌,滴加饱和naoh溶液至ph=10,将固体过滤,滤饼用冰水(50ml)洗涤,干燥得到式5所示化合物,得量13.40g,收率66.1%。
29、根据本发明的实施例,在步骤(4)中,式5所示化合物与式6所示化合物的接触方式并不受特别限制。由此,可以提升式5所示化合物与式6所示化合物接触反应的效率,加快反应速度,进一步提高利用该方法制备式7所示化合物的效率。
30、根据本发明的实施例,在步骤(4)中,包括如下步骤:室温下,将式5所示化合物加入短链醇a中,加入式6所示化合物,然后升温至回流状态,搅拌反应3小时~3小时30分钟,tlc监测反应完成,反应液降至室温,过滤,滤饼用冰水洗涤,干燥得到式7所示化合物。由此,可以提升式5所示化合物与式6所示化合物接触反应的效率,加快反应速度,进一步提高利用该方法制备式7所示化合物的效率。
31、根据本发明的实施例,在步骤(4)中,式5所示化合物与式6所示化合物的摩尔比为1:(2.5~4.0),优选式5所示化合物与式6所示化合物的摩尔比为1:3.0。由此,可以进一步提高利用该方法制备式7所示化合物的效率。
32、根据本发明的实施例,在步骤(4)中,短链醇a为选自异丙醇、或乙醇的至少一种。
33、根据本发明的实施例,在步骤(4)中,优选搅拌反应3小时15分钟。
34、根据本发明的一个具体实施例,在步骤(4)中,包括如下步骤:室温下,将式5所示化合物(10.0g,34.16mmol)加入异丙醇(100ml)中,加入式6所示化合物(13.86g,102.48mmol),然后升温至回流状态,搅拌反应3小时15分钟,tlc监测反应完成,反应液降至室温,过滤,滤饼用冰水(50ml)洗涤,干燥得到式7所示化合物,得量12.40g,收率92.7%。
35、根据本发明的实施例,在步骤(5)中,式7所示化合物与式8所示化合物、磷酸钾、碘化物的接触方式并不受特别限制。由此,可以提升式7所示化合物与式8所示化合物、磷酸钾、碘化物接触反应的效率,加快反应速度,进一步提高利用该方法制备式i所示化合物的效率。
36、根据本发明的实施例,在步骤(5)中,包括如下步骤:室温下,将式7所示化合物,式8所示化合物,磷酸钾和碘化物混溶于干燥的2-丁酮中,反应液在氮气氛围中,加热保持回流状态,搅拌反应17小时~20小时,反应结束后,冷却至室温后用硅藻土过滤,滤饼用二氯甲烷洗涤,无水硫酸钠干燥,滤液减压浓缩蒸出溶剂,浓缩物用二氯甲烷/甲醇混合溶剂,经硅胶柱层析纯化,得到式i所示化合物恩塞芬汀。由此,可以提升式7所示化合物与式8所示化合物、磷酸钾、碘化物接触反应的效率,加快反应速度,进一步提高利用该方法制备式i所示化合物恩塞芬汀的效率。
37、根据本发明的实施例,在步骤(5)中,式7所示化合物与式8所示化合物、磷酸钾、碘化物的摩尔比为1:(2.0~4.0):(4.0~6.0):(2.0~4.0),优选式7所示化合物与式8所示化合物、磷酸钾、碘化物的摩尔比为1:3.0:5.0:3.0。由此,可以进一步提高利用该方法制备式i所示化合物的效率。
38、根据本发明的实施例,在步骤(5)中,所述碘化物为选自碘化钠、或碘化钾的至少一种,优选碘化物为选自碘化钠。
39、根据本发明的实施例,在步骤(5)中,所述二氯甲烷/甲醇混合溶剂中二氯甲烷与甲醇的体积比为(15~25):1,优选二氯甲烷与甲醇的体积比为20:1。
40、根据本发明的实施例,在步骤(5)中,优选搅拌反应18小时。
41、根据本发明的一个具体实施例,在步骤(5)中,包括如下步骤:室温下,将式7所示化合物(10.0g,25.55mmol),式8所示化合物(12.80g,76.64mmol),磷酸钾(27.11g,127.73mmol)和碘化钠(11.49g,76.64mmol)混溶于干燥的2-丁酮(300ml)中,反应液在氮气氛围中,加热保持回流状态,搅拌反应18小时,反应结束后,冷却至室温后用硅藻土过滤,滤饼用二氯甲烷(100ml)洗涤,无水硫酸钠干燥,滤液减压浓缩蒸出溶剂,浓缩物用体积比为20:1的二氯甲烷/甲醇混合溶剂,经硅胶柱层析纯化,得到式i所示化合物恩塞芬汀,得量9.44g,收率77.4%,hplc纯度99.6%。
42、根据本发明的具体实施例,式i所示化合物恩塞芬汀的合成路线可以如下所示:
43、
44、相对于现有技术,本发明所述的恩塞芬汀的制备方法,其至少具有以下有益效果:
45、1、相对于现有技术,本发明所述制备方法的有益效果在于:本方法将初始原料式1所示化合物和式2所示化合物经过sizuki偶联反应得到式3所示化合物,经过分子内成环得到式4所示化合物,利用pocl3将羰基转化为卤化物(式5所示化合物),再与式6所示化合物经过烷基化反应得到式7所示化合物,其中式7所示化合物的酰基亚胺再与溴化脲发生取代反应得到产物恩塞芬汀。
46、2、本发明与现有技术方法相比,其显著优点在于:(1)wo2012037782面对带有羟基的化合物时,先将羟基保护后再发生取代反应,本发明选择硼酸化合物作为起始原料,与化合物发生sizuki反应,无需对羟基进行保护就能获得较高的反应产率,减少了羟基的保护与脱保护共两步反应。(2)wo2023109802文献中将式7所示化合物和2-(2-溴乙基)异-哚啉-1,3-二酮反应得到易脱去化合物,再加入水合肼得到胺基,加入氯酸钾得到终产物(脲)。本发明直接将式7所示化合物与卤代脲反应得到终产物,缩短了2步反应、简化了反应操作。(3)本发明选用的原料易得,操作方法简便,产品产率较高,适合批量化生产。
本文地址:https://www.jishuxx.com/zhuanli/20250110/353904.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表